

概要
ゲルろ過カラムからの溶出体積を基にタンパク質の分子量を見積もる方法 (1) は、簡便な分子量分析手法として頻用されている。だが解析対象となる分子がゲルろ過担体と相互作用する条件下や、解析対象となる分子の形状が球状からかけ離れている場合では、真の分子量とゲルろ過カラムからの溶出体積から見積もられる分子量に大きな違いが認められる。ところがそのような条件下においても、ゲルろ過カラムから溶出したタンパク質の静的光散乱と濃度を測定することによって、溶出体積を基にした分析よりも、原理上信頼性の高い分析結果を得ることができる (2,3)。溶出体積を基にした分析法との実験的な違いは、カラムから溶出したタンパク質の検出法だけであり、実験にかかる時間や必要な試料の量や純度等には違いはない。ゲルろ過カラムクロマトグラフィーと静的光散乱検出器および示差屈折率計を組み合わせた分子量分析法はとりたてて新規な手法ではないが、測定装置の改良が進んで比較的容易にかつ再現性よく実験を行えるようになったため、ここであらためて紹介したい。本方法を用いて、これまで安定だと考えられていたヒストン (H3-H4)2 四量体が、内在性因子(ヒストンシャペロン CIA)によって二つの二量体に分割されることが明らかになった (4)。
イントロダクション
解析対象となるタンパク質が単量体なのか、あるいは同一のポリペプチド複数からホモ多量体を形成するのか、それとも異種ポリペプチドでヘテロ複合体を形成するのか、またホモ多量体やヘテロ複合体を形成する場合の化学量論はどうなっているのか、分子の会合状態に関する情報は、タンパク質の機能を解析する上で極めて基本的な情報である。分子の会合状態は、多くの場合において分子量から求めることが可能である。通常タンパク質の分子量分析は、超遠心分析、動的光散乱、ゲルろ過カラムクロマトグラフィー、SDS-PAGE、Native-PAGE 等のいずれかの手法単独で、あるいはこれらの手法を複数組み合わせて行われる。なかでも、ゲルろ過カラムクロマトグラフィーの溶出体積から分子量を見積もる方法 (1) は簡便なため、頻用されてきた。しかし、タンパク質分子がゲルろ過担体と相互作用する場合(塩基性タンパク質の場合しばしば観察される)や、タンパク質分子の形状がモデルとなる球状タンパク質と大きく異なる場合においては、溶出体積から “正しい” 分子量を見積もることは困難である。
ところが、ゲルろ過カラムから溶出するタンパク質の検出に静的光散乱検出器と示差屈折率計を用いれば、簡便性というゲルろ過カラムクロマトグラフィーの利点を活かしつつ、上記のような欠点を克服してより正確に分子量を分析することができる (5,6)。ゲルろ過カラムクロマトグラフィーでは検出部に吸光度計が用いられることが一般的であるが、本方法では検出部に静的光散乱検出器と示差屈折率計を用いる(図1)。静的光散乱検出器では溶出タンパク質の分子量および濃度の両者に比例する値が、示差屈折率計では溶出タンパク質の濃度に比例する値が測定できる。理論的な側面についてはいろいろな総説等 (2,3,7) で解説されているが、以下に概説する。
溶質の濃度が希薄な条件下では、光散乱強度、散乱角度、溶質の分子量、溶質濃度の基本関係は、以下の式で表される。
\[\frac{K^{*}c}{R(\theta)} = \frac{1}{M_{w}} \left[ 1 + \frac{16\pi^{2}}{3\lambda^{2}} \langle {r_{g}}^{2} \rangle \sin^{2}(\theta / 2) + \cdots \right] + 2A_{2}c \tag{1}\]ここで、\(R(\theta)\) は角度 \(\theta\) における過剰散乱強度の還元散乱強度、\(K^{*}\) は光学パラメーターで \(4\pi^{2}{n_{0}}^{2}(dn/dc)^{2}/({\lambda_{0}}^{4}N_{A})\)、\(c\) は \(\mathrm{mg{\ }ml}^{-1}\) を単位とする溶質の濃度、\(n_{0}\) は溶媒の屈折率、\(dn/dc\) は溶質の屈折率の濃度増分、\(\lambda_{0}\) は真空中での入射光の波長、\(N_{A}\) はアボガドロ数、\(M_{w}\) は重量平均分子量、\(\langle {r_{g}}^{2} \rangle\) は平均二乗慣性半径、\(A_{2}\) は溶質分子の分散状態(溶質分子と溶媒分子の親和性)を示す第二ビリアル係数である。ゲルろ過カラムクロマトグラフィーからの溶出液などの溶質の濃度が希薄な条件下では、第二ビリアル係数を無視してもよい。またおおまかな目安として、溶質の根平均二乗慣性半径 \({\langle {r_{g}}^{2} \rangle}^{1/2}\) が静的光散乱の測定波長の1/20以下の場合には、散乱光の角度依存性はほとんどないものとして扱ってよいとされており、その場合、式(1)の角度依存性を表している角度関数の項 \([ 16\pi^{2} \langle {r_{g}}^{2} \rangle \sin^{2} (\theta / 2) ] / ( 3\lambda^{2} )\) を無視することができるようになる。つまり、大抵のタンパク質をゲルろ過カラムクロマトグラフィーから溶出させた際の、光散乱強度、溶質の分子量および濃度の基本関係は、式(1)より \(M_{w} = R(\theta) / (K^{*}c)\) の式で近似してもよいことになり、タンパク質の分子量は、タンパク質の濃度に対する散乱光の強度の比から求められることになる。静的光散乱検出器のレーザー光源の特性上、測定には 633 nm あるいは 690 nm 付近の波長が用いられることが多いため、ここでは根平均二乗慣性半径 \({\langle {r_{g}}^{2} \rangle}^{1/2}\) が 30 nm を超える様な “大きな” タンパク質の分析は想定していない。この場合、散乱光はどの角度で測定してもよいが S/N 比が最も高い 90° で行うとよいだろう。なお、Kaye らが開発した小角での散乱光を測定するタイプの静的光散乱検出器 (8) を用いる場合には、ここで想定しているよりも “大きな” タンパク質を測定する場合においても式(1)を \(M_{w} = R(\theta) / (K^{*}c)\) と近似できるため、本プロトールによる分析も可能である。本プロトコールの範疇を超えるような “大きな” タンパク質の分析を行うには、複数の角度で散乱光を検出することができる多角度光散乱検出器を使用し、散乱光強度の角度依存性を用いて分子量を求める方法 (7) の方がよいだろう。記録される静的光散乱検出器の出力強度 \(Int_{\mathrm{LS}}\) は、\(K_{\mathrm{LS}}\) を静的光散乱測定の装置定数(使用する測定装置および測定条件に応じて決まる値)とすると、溶質の分子量および濃度の両者に比例する値として次式(2)で表すことができる。
\[Int_{\mathrm{LS}} \approx K_{\mathrm{LS}}cM_{w}(dn/dc)^{2} \tag{2}\]また、\(K_{\mathrm{RI}}\) を示差屈折率測定の装置定数とすると、示差屈折率計の検出強度 \(Int_{\mathrm{RI}}\) は、溶質の濃度 \(c\) に比例する値として次式(3)で表すことができる。
\[Int_{\mathrm{RI}} \approx K_{\mathrm{RI}}c(dn/dc) \tag{3}\]ゲルろ過カラムクロマトグラフィーの検出部に、静的光散乱測定器と示差屈折率計を直列に接続して、溶出タンパク質の示す静的光散乱検出器の強度と示差屈折率計の強度を測定すると、式(2)および式(3)の左辺の値が実測値として得られる。式(2)および式(3)から(\(c\) を消去する事で)、重量平均分子量 \(M_{w}\) と実測値の関係が次式(4)のように求められる。
\[M_{w} \approx \frac{K_{\mathrm{RI}}}{K_{\mathrm{LS}}(dn/dc)} \frac{Int_{\mathrm{LS}}}{Int_{\mathrm{RI}}} = K'\frac{Int_{\mathrm{LS}}}{Int_{\mathrm{RI}}} \tag{4}\]一般的に、大腸菌で発現させた組換えタンパク質の様な糖鎖修飾を持たないタンパク質に関しては、屈折率の濃度増分 \(dn/dc\) の値はアミノ酸配列に依存せず約 \(0.186{\ }\mathrm{g^{-1}{\ }ml}\) と一定であるとしてよいとされている (2,3)。そのため、分子量が既知の標準タンパク質を複数用いて静的光散乱検出器と示差屈折率計の出力強度を測定し、分子量を溶出ピーク頂点の \(Int_{\mathrm{LS}} / Int_{\mathrm{RI}}\) の値に対してプロットして検量線を作製すれば、その傾きから静的光散乱と示差屈折率を同時に測定する際の装置定数 \(K' = K_{\mathrm{RI}} / [ K_{\mathrm{LS}} (dn/dc) ]\) を比較的精度よく求められる。標準タンパク質には、糖鎖修飾を受けていないもの(あるいは糖鎖修飾を受けていてもごくわずかで、糖鎖修飾の \(dn/dc\) への影響が少ないことが分かっているもの)で、分子量が既知のものであれば何を用いても特に問題はない。用いる分析装置の装置定数 \(K’\) が実験的に求まれば、式(4)によって、解析対象となるタンパク質の分子量を見積もることができる。本プロトコールによる分子量分析は、以上のように行われる。なお、糖鎖修飾によって \(dn/dc\) 値は影響を受けるため、糖鎖修飾を持つタンパク質の分子量分析には本プロトコールをそのまま適用することはできない。
静的光散乱検出器と示差屈折率計の測定強度は、溶出タンパク質のピークの高さから求めても面積から求めてもどちらでも基本的に問題はないが、ピークの高さから求める方が簡便である(図2)。筆者らは、外部入力を取り込む A/D コンバータを内蔵した、コンピューター制御の静的光散乱検出器を用いているため、クロマトグラムのテキストデータから測定強度をそのまま読み取り、ピークの高さを得ている。各検出器からの出力をチャート紙で記録して紙媒体上でピークの高さを読み取っても大抵の場合分析は成立するが、Smart Chrom(KYA TECH 社)等の A/D コンバータを用いて測定強度を数値化した方が解析しやすいだろう。
本プロトコールでは吸光度計の出力強度を分析には用いていないが、扱うタンパク質の分子吸光係数 \(\varepsilon\) が既知であれば、吸光度計の出力強度を用いても同様の分析が可能である。\(K_{\mathrm{UV}}\) を吸光度測定の装置定数、\(\varepsilon\) を \(\mathrm{mg^{-1}{\ }ml{\ }cm^{-1}}\) を単位とする分子吸光係数とすると、吸光度計の出力強度 \(Int_{\mathrm{UV}}\) を次式(5)で表すことができ、式(2)および式(5)から重量平均分子量 \(M_{w}\) と実測値の関係式(6)が求められる。
\[Int_{\mathrm{UV}} = K_{\mathrm{UV}}c\varepsilon \tag{5}\] \[M_{w} \approx \frac{K_{\mathrm{UV}}}{K_{\mathrm{LS}}(dn/dc)^{2}} \varepsilon \frac{Int_{\mathrm{LS}}}{Int_{\mathrm{UV}}} = K'' \varepsilon \frac{Int_{\mathrm{LS}}}{Int_{\mathrm{UV}}} \tag{6}\]\(dn/dc\) 値を約 \(0.186{\ }\mathrm{g^{-1}{\ }ml}\) と一定であるとして扱える場合は、式(4)を用いる本プロトコールの方法と同様に標準タンパク質を用いて検量線を作製して \(K''\) を求めれば、式(6)を用いて分析が成立する。大腸菌で発現させた組換えタンパク質で、単一種類のサブユニットからなるタンパク質(複合体)が解析対象であれば、アミノ酸配列から算出した分子吸光係数を用いて解析を行っても特に問題はないだろう。だが複数種類のサブユニットからなる複合体の場合には、サブユニット構成比があらかじめ分かっていなければ分子吸光係数 \(\varepsilon\) を算出できず、測定強度から直接的に分子量を求めることができない。そのために本プロトコールでは吸光度計の測定強度を用いた分析方法についてはとりあげないことにした。だが吸光度は分子種や純度を知る手掛かりになるため、検出部には吸光度計も加えておくとよいだろう。吸光度も測定しておくと、\(dn/dc \approx 0.186{\ }\mathrm{g^{-1}{\ }ml}\) と扱える場合には、式(3)および式(5)から導かれる次式(7)を用いて分子吸光係数 \(\varepsilon\) を算出することも可能になる。
\[\varepsilon \approx \frac{(dn/dc)K_{\mathrm{RI}}}{K_{\mathrm{UV}}} \frac{Int_{\mathrm{UV}}}{Int_{\mathrm{RI}}} \tag{7}\]分子吸光係数 \(\varepsilon\) があらかじめ別の方法で決定されていなければ分析が成立しないため本プロトコールではとりあげないが、\(dn/dc\) 値を一定として扱えない糖鎖修飾を持つタンパク質の場合でも、吸光度計の測定強度を用いることで、式(2)、式(3)、式(5)から導かれる次式(8)を用いて、分子量分析を行うことが可能であることも付け加えておく。
\[M_{w} \approx \frac{(K_{\mathrm{RI}})^{2}}{K_{\mathrm{LS}}K_{\mathrm{UV}}} \frac{Int_{\mathrm{LS}} Int_{\mathrm{UV}}}{(Int_{\mathrm{RI}})^{2}} \frac{1}{\varepsilon} \tag{8}\]また、本プロトコールのようにゲルろ過カラムを用いなくても、試料を直接検出器に打ち込んで測定する方法で原理的には式(4)によって重量平均分子量を求めることも可能である。だが、タンパク質溶液の溶媒を完全に置換しておき溶液中のタンパク質の屈折率のみを実験的に測定することが非常に難しい。分析試料中の侠雑タンパク質や強い光散乱を示す高分子凝集成分等を解析対象のタンパク質から分離する目的も兼ねているが、分析タンパク質試料の溶媒を完全に置換して溶出タンパク質の屈折率をきちんと測定する目的で、本プロトコールではゲルろ過カラムを用いている。ただし、ゲルろ過カラムを用いると、試料の希釈が必ず起きてしまうため、\(K_{\mathrm{D}}\) が μM オーダーの比較的弱い相互作用によって形成されているような複合体試料の分析には本プロトコールの分析方法は不向きであることに留意されたい。なお、ゲルろ過カラムを用いる分析全般に言えることであるが、高純度なタンパク質試料を準備すること、カラムの種類、カラムのサイズ、ランニングバッファーの組成、流速等の条件をあらかじめ検討し、解析対象となるタンパク質試料がなるべく鋭くかつ左右対称なピークとして溶出する高分離な条件を確立しておくことが肝要である。
装置・器具・試薬
- ランニングバッファー(条件に応じて調製)
- PEEK製チューブおよびコネクター(各社)
- 送液ポンプ(RID-10A,(株)島津製作所)*
- PEEK製インラインフィルターホルダー(Wyatt technology)*
- インラインフィルターメンブレンディスク(ポアサイズ 0.1 μm、直径 25 mm、各社)
- インジェクションバルブ(9725i、RHEODYNE)*
- サンプルループ(各社)
- ガスタイトシリンジ(各社)
- ゲルろ過カラム(各社)
- 吸光度計(SPD-20A,(株)島津製作所)*
- 静的光散乱検出器(miniDAWN tristar,Wyatt technology)*
- 示差屈折率計(RID-10A,(株)島津製作所)*
- レコーダー(各社)
- 標準タンパク質(RNase,ovalbumin,BSA 等)
- 解析対象となるタンパク質(なるべく高純度に精製したもの)
* 筆者らが用いている機器名および製造元を示してある。
送液ポンプ;0.1 ml/min 程の低流速での送液が安定に行えるものであれば、使用できる。一般的な HPLC 用のポンプであればまず問題ない。単体で動作させるだけなので、HPLC システム用のコントローラー等は必要ない。筆者らは送液部のコントロールをマニュアルで行っている。
静的光散乱検出器;フローセルで測定できる機器にはあまり選択肢がないのが実情である。できるだけフローセル容量の小さな機器が望ましい。小角専用タイプ (8) であればフローセル容量が小さいが、検出感度が高い一方で測定のバックグラウンドが高くなりやすい。筆者らには経験がないが、フローセル容量が小さな HPLC 用の蛍光検出器であれば、本プロトコールに流用できる可能性がある。また、上市されているかどうかの情報を筆者らはもちあわせていないが、吸光度と 90° の散乱光を同時に測定できるタイプのフローセルを備えた機器も開発されている (9)。
示差屈折率計;静的光散乱検出器で用いる測定波長と同じ波長で測定できるタイプの機器が理想的であるが、筆者らが用いている測定波長の設定ができないタイプの機器でも特に問題はない。
実験手順
- 分析条件の確立
- 実験装置、カラムの平衡化
- ゲルろ過カラムのクロマトグラム測定
- クロマトグラムデータの解析
- 装置定数の決定
- 解析対象分子の分子量決定
実験の詳細
1. 分析条件の確立
解析対象の試料はなるべく高純度に、かつ大量に精製する。分子量分析に適した条件は、解析対象となるタンパク質試料がなるべく鋭く、かつ左右対称なピークとして溶出する高分離が可能な条件である。そのため、あらかじめ、カラムの種類、カラムのサイズ、ランニングバッファーの組成、流速等の条件を検討し、分子量分析に適した条件を確立する。
2. 分析装置、カラムの平衡化
ランニングバッファーはメンブレンフィルターを用いてあらかじめ濾過しておき、流路で気泡が発生しないように脱気も十分行っておく。可能であれば、ランニングバッファーに He ガスを通気しておくと良い。上記1)で確立した条件下で、実験装置にカラムを接続し、終夜平衡化する。示差屈折率計の流路は、リファレンスセルとサンプルセルの両方にランニングバッファーが流れるように設定しておく。本プロトコールでは、装置定数 \(K_{\mathrm{LS}}\)、\(K_{\mathrm{RI}}\) をそれぞれ個別にキャリブレーションする必要はない。
3. ゲルろ過カラムのクロマトグラム測定
サンプルセルのみにランニングバッファーが流れるように示差屈折率計の流路を設定し、標準タンパク質の測定と、解析対象のタンパク質の測定を、それぞれ3回以上行う。示差屈折率計の測定は特に温度変化に対して敏感なため、測定は同じタンパク質試料に関して複数回連続して行うのではなく、測定装置の状態が一連の測定を通して一定であることを確かめられるように、例えば標準タンパク質と解析対象のタンパク質の測定を交互に行った方がよい。測定の合間には、示差屈折率計のフローセルの流路を切り換えてリファレンスセルの平衡化を行うとよい。標準タンパク質を含めた一連の測定を一定の条件で行うためには途中でランニングバッファーを交換すべきではないため、十分量のランニングバッファーを用意しておき、一連の測定をなるべく短期間(できれば1~2日)のうちに終えてしまう方がよい。測定強度が検出器のダイナミックレンジを超えてしまった場合には、より低い濃度の試料で測定をやり直す。
Wyatt Technology の製品を用いる場合;付属のソフトウェアに測定時の流速値を入力し、試料のインジェクトと同時に測定を開始する。測定を開始すると、各検出器のクロマトグラムは電圧変化の値として記録され、設定した時間(30分前後)で1回の測定が完了する。静的光散乱検出器と示差屈折率計で検出される溶出ピークの体積ギャップのキャリブレーションは記録される生データには反映されないため必ずしも必要ではないが、最初に BSA 等の標準タンパク質を用いて決定し、付属のソフトウェアに入力しておくとよい。
4. クロマトグラムデータの解析
各クロマトグラムから、検出強度のベースラインに対する溶出ピークの高さ(静的光散乱検出器の測定強度 \(Int_{\mathrm{LS}}\) と示差屈折率計の測定強度 \(Int_{\mathrm{RI}}\))を読み取る(図2)。
Wyatt Technology の製品を用いる場合;付属のソフトウェアを用いて各クロマトグラムのベースラインの値を差し引き、テキストデータとして保存する。なお、以降の作業では付属のソフトウェアを使う必要はない。保存されたテキストデータから 90° の光散乱検出器の出力ピーク値、示差屈折率計の出力ピーク値を読み取る。また、ピークの溶出体積も読み取っておき、あらかじめ求めておいた検出器間のギャップと一致していることを確認しておく。分析試料データの溶出ピークから算出される検出器間のギャップが標準タンパク質のそれと異なる場合は、試料の分離がうまくいっていない可能性が高いため、分析条件を再検討すべきであろう。
5. 装置定数の決定
標準タンパク質の \(Int_{\mathrm{LS}} / Int_{\mathrm{RI}}\) の平均値を用いて検量線を作成する(図3)。原点を通る直線の傾きから、式(4)の装置定数 \(K' = K_{\mathrm{RI}} / [ K_{\mathrm{LS}} (dn/dc) ]\) を求める。
6. 解析対象のタンパク質の分子量決定
式(4)を用い、解析対象タンパク質の \(Int_{\mathrm{LS}} / Int_{\mathrm{RI}}\) と上記 5. で求めた装置定数 \(K' = K_{\mathrm{RI}} / [ K_{\mathrm{LS}} (dn/dc) ]\) から、解析対象タンパク質の重量平均分子量を見積もる。
工夫とコツ
分析条件検討時には吸光度のみを検出すればよい
吸光度計のみで溶出パターンの確認はできるため、分析条件を検討する際には、ゲルろ過カラムクロマトグラフィーシステムの検出部から、静的光散乱検出器と示差屈折率計を取り外しておいた方がよい。条件によっては、解析対象のタンパク質がゲルろ過カラムに吸着してしまい、アルカリや酸を用いてカラムの洗浄操作を行わなければならない場合もあり、その洗浄操作によって溶出した成分が静的光散乱検出器のフローセル内部に吸着して感度の高い静的光散乱測定のノイズが大きくなってしまうことを避けるためでもある。筆者らは、静的光散乱検出器を接続したままカラム洗浄を行ってしまい、静的光散乱検出器のフローセルについた汚れを除去するのに苦労した経験がある。カラムの洗浄操作には、分析用のものとは別のクロマトグラフィーシステムを用いることが望ましい。
ランニングバッファーによる装置の腐食を防ぐ
タンパク質試料とカラムの相互作用を低減させる目的で、ある程度の濃度の塩(通常 0.1 M 以上)を含むランニングバッファーを用いることが多いが、金属製のジャケットのカラムを用いる場合には、金属の腐食を避けるために、可能な限りハロゲンを含まない硫酸ナトリウム等の塩を用いた方が安全である。ハロゲンを含む塩を用いる場合、ポンプとインジェクションバルブの間に接続するインラインフィルターホルダーやインジェクションバルブ等、流路に接続するものはできるだけ PEEK 製のものが望ましい。また、プランジャー式のポンプの場合、プランジャーの腐食を防ぐために、専用のプランジャー洗浄ユニットをポンプに装備するか、ペリスタポンプを用いてプランジャーに水を送液しつづけるとよい。筆者らは、金属製のインラインフィルターホルダーや洗浄ユニットを装備していない状態で使っていたポンプのプランジャーにさびが発生し、メンテナンスに苦労した経験もある。
測定温度
実験装置にカラムを接続した後に終夜平衡化する目的は、ランニングバッファーを含む実験装置全体の温度を平衡にするためである(特に屈折率は温度の変化に敏感である)。示差屈折率計の多くは、温度をコントロールするためにヒーターを内蔵しているものの冷却機能を内蔵していないため、分析を行う温度よりも室温を低く設定した部屋に分析装置を設置するか、装置内蔵の温度コントロール機能を使わずに室温が一定の部屋に設置するとよい。
実験スケールと検出器の接続について
分析用途として販売されている小さいカラムを用いると数μlほどの少量の試料でも分析が可能であるが、分析精度が低くなることに注意した方が良い。カラムから溶出するタンパク質の体積が静的光散乱検出器のフローセル容積(筆者らが用いている静的光散乱検出器のフローセル容量は 80 μl)に対して小さくなりすぎてしまい、その結果、静的光散乱検出器のフローセル内部における希釈の影響が大きくなり、静的光散乱が検出される際の溶出タンパク質の濃度と、示差屈折率計で測定される際の溶出タンパク質の濃度の違いが大きくなってしまうからである。静的光散乱と示差屈折率を測定する際の溶出タンパク質の濃度がほぼ一定に保たれた良質のデータを得るには、ある程度の大きさのカラム(内径 5 mm 以上*長さ 200 mm 以上)を用いた方が良い。通常分取に用いられる内径 10 mm*長さ 300 mm のカラムを用いた場合でも、一回の測定に数 mg/ml 以上の濃度のタンパク質試料を 20 μl ほど用いれば、良質のデータを得られる。カラムサイズの違いによる分析精度の違いを、一例として示す (4)(表1)。なお、静的光散乱検出器のフローセル中における溶出タンパク質の希釈の影響は、実験的に求められる装置定数 \(K'\) に含まれた形で補正されることになる。フローセル容積の小さな示差屈折率計を流路の上流側に、フローセル容積の大きな静的光散乱検出器を流路の下流側に接続すれば、静的光散乱検出器のフローセル内部における溶出タンパク質の希釈は問題にならなくなるが、そのような接続は示差屈折率計の背圧を高めフローセルの破損をまねくことになるためさけるべきである。一般的に示差屈折率計のフローセルは耐圧限界が低いため、安全のために示差屈折率計は流路の最も下流側に設置した方がよい。静的光散乱検出器と示差屈折率計を流路上に直列に接続するのではなく、カラムからの溶出液を2つの流路に分けて静的光散乱検出器と示差屈折率計を並列に接続した場合には、もっと小型のカラムを用いてもきれいなデータを得ることができると考えられる。ただしその場合、内径が非常に小さな PEEK チューブを流路の廃液側に接続するなどして2つの検出器の背圧をコントロールし、各検出器の流量を一定に保つ工夫が必要になるだろう。
インジェクションされる試料の量をコントロールする
複数回の分析で再現性の高い結果を得るには、毎回同じ量の試料をカラムにインジェクトするべきである。用いるサンプルループの容量が例えば 20 μl だった場合、30 μl の試料をシリンジに吸い込み、サンプルループには 25 μl 強の試料を打ち込むようにして、サンプルループを試料で完全に満たすようにするとよいだろう。表1において、カラム2で求めたヒストン (H3-H4)2 四量体の分子量の S.D. 値が大きい(とはいっても ±1,050 Da である)のは、サンプルループにロードした試料の量が、サンプルループの容量の約半分だった際のクロマトグラムのデータも解析に用いたためである。
分析法の比較
溶出体積の記録も精密に行える場合、本プロトコールによる分析だけではなく、ピークの溶出体積から分析する方法 (1) でも分子量を求めてみるとよい。一例を表2に示す。ヒストン H2A-H2B 二量体やヒストン (H3-H4)2 四量体の場合、本プロトコールによって見積もられた分子量の方が理論値に近く、溶出体積から求められる分子量は理論値よりもかなり大きめに見積もられていることが分かる。2つの方法で分析すれば、各ヒストンサブユニットの N 末端領域に存在する tail 領域が定まったフォールドをとっていないために、両ヒストン複合体分子全体の形状が標準タンパク質と比較して非対称的になっており、このような結果になったと考えることも可能である。
測定装置のメンテナンス
装置の使用後は、脱気した水で十分に洗ってから、100%メタノールでさらに十分洗うとよい。流路から検出機器を外しておく場合は、さらにイソプロパノール:アセトニトリル:水=40%:40%:20% (v/v) 混合液でフローセルを満たし、同じ混合液で満たした PEEK チューブで検出器の IN と OUT を接続しておくとよい。
タグ融合型タンパク質の化学量論を分析する際は要注意
最近ではほとんどの組換えタンパク質の精製にアフィニティータグが利用されている。解析対象のタンパク質を簡便に精製することができる利点はあるが、タグによって分子の会合状態に影響がでる場合があるので、タグ融合型タンパク質の化学量論を分析する際には注意が必要である。文献4には記載しなかったが、N 末のヒスチジンタグを除去していないタグ融合型 CIA の場合、単量体と二量体の2つのピークが観察されたのである。N 末のヒスチジンタグを除去すると二量体のピークは観察されなくなり、単量体のピークのみが観察された。タグ融合型タンパク質を用いて化学量論を分析する際には、タグを除去したタンパク質と化学量論に違いがないかどうか、確認すべきであろう。
文献
- Handbook Gel Filtration Principles and Methods. GE healthcare (2002).
- Takagi, T. J. Chromatogr., 506, 409–416 (1990).
- Wen, J., Arakawa, T. & Philo, J. S. Anal. Biochem., 240, 155–166 (1996).
- Natsume, R., Eitoku, M., Akai, Y., Sano, N., Horikoshi, M. & Senda, T. Nature, 446, 338–341 (2007).
- Fukuda, M., Jukutomi, M. & Hashiomoto, T. Polymer Preprints, Jpn., 28, 1646–1650 (1979).
- Takagi, T. J. Biochem., 89, 363–368 (1981).
- Wyatt, P.J. Anal. Chim. Acta., 272, 1–40 (1993).
- Kaye, W., Hvlik, A. J. & McDaiel, J. B. J. Chromatogr., 9, 695–699 (1971).
- Bajaj. H., Sharma. V. K. & Kalonia. D. S. Biophys. J., 87, 4048–4055 (2004).
-
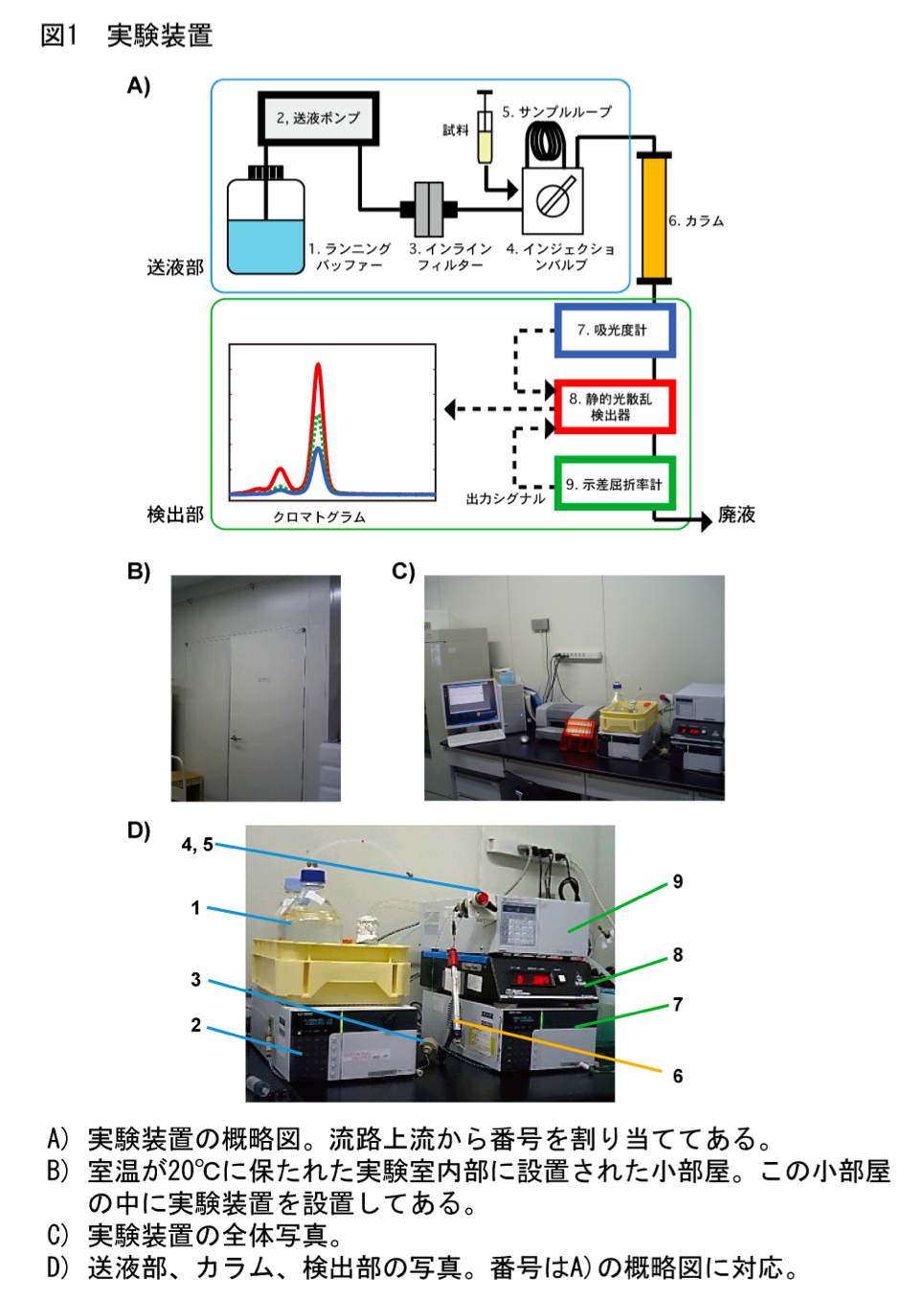
図1: -
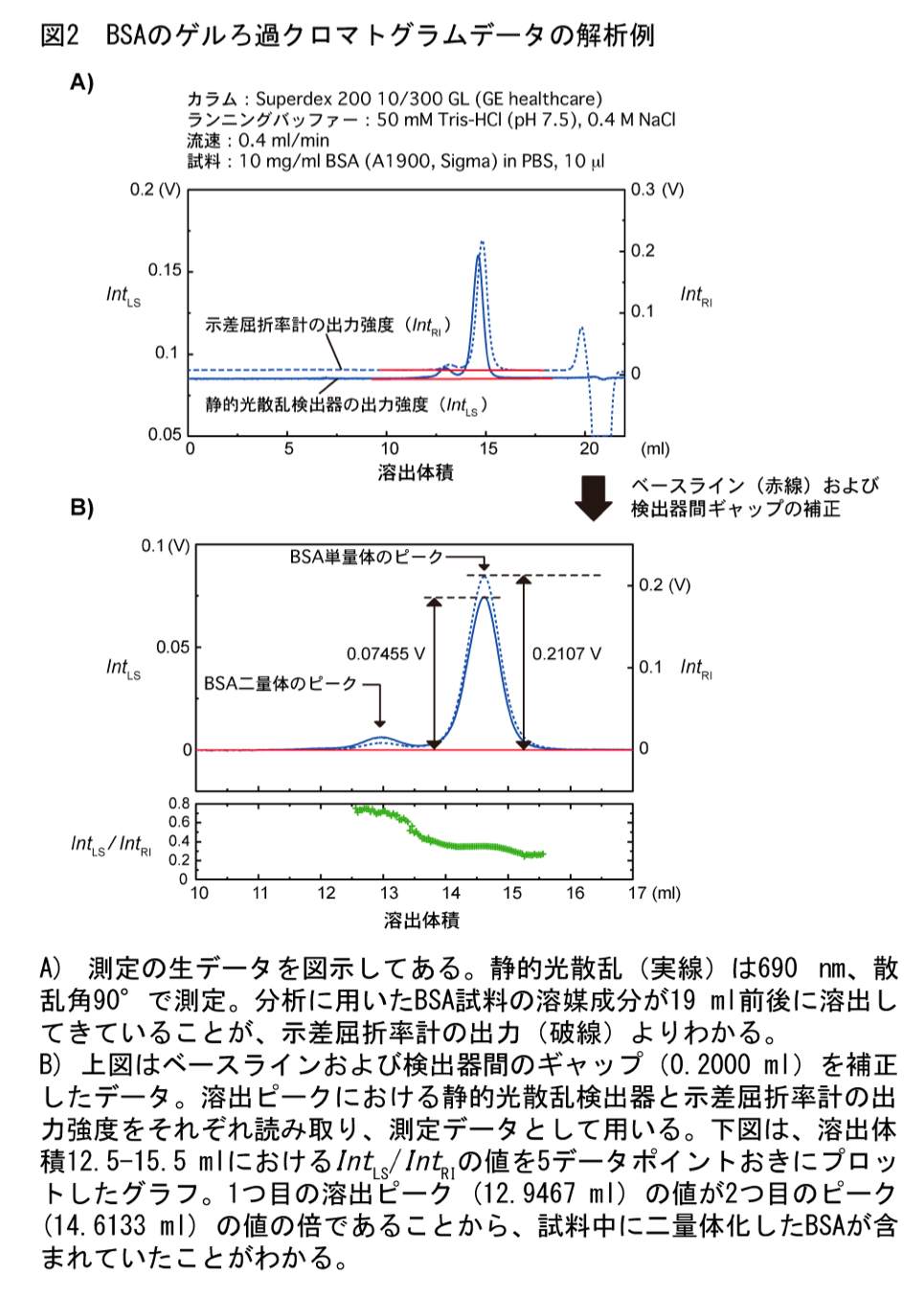
図2: -

図3: -

表4: -
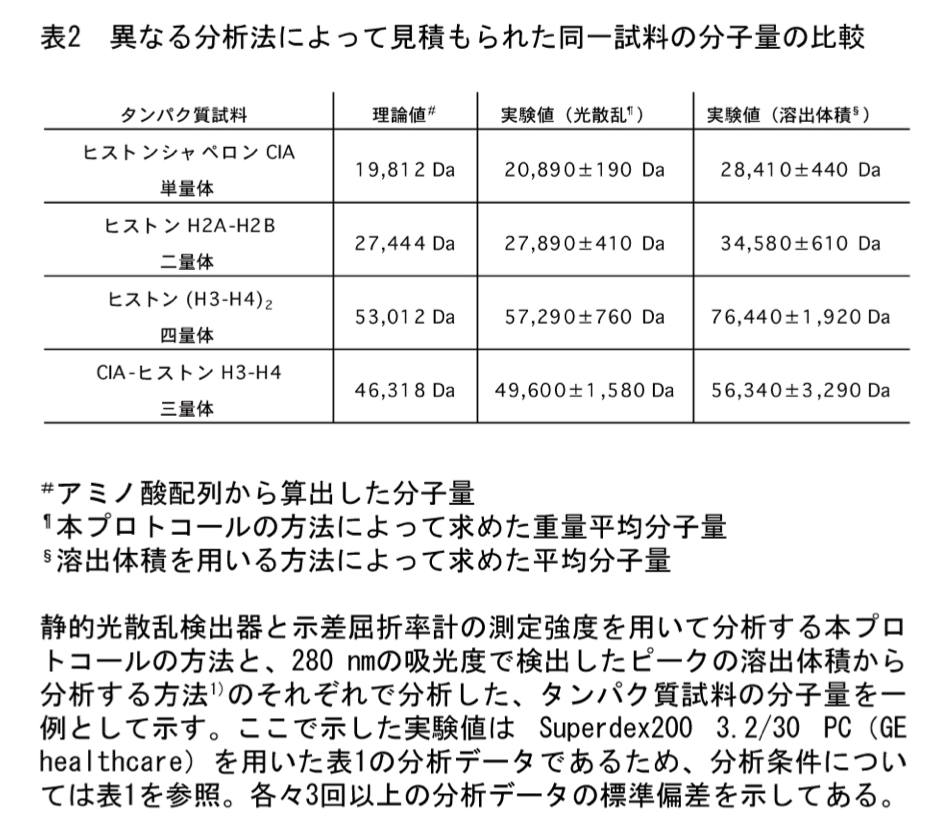
表5:
