

概要
X線結晶構造解析において、タンパク質の立体構造を求めるためには、まず目的タンパク質の結晶が必要である。そして、その結晶を用いてX線回折実験を行い、X線回折強度データを収集する。ただし、X線回折強度データが得られればすぐに構造解析ができるかというとそうではない。構造解析には、X線回折強度データに加え、位相情報が必要となるからである。現在、タンパク質のX線結晶構造解析で位相を求める方法として、分子置換法(MR法)、重原子同型置換法(MIR法)、多波長/単波長異常分散法(MAD/SAD法)といった方法がよく用いられている。この中で、MR法は、既知の類似タンパク質の構造とX線回折強度データを用いて位相を決定する比較的簡便な手法である。一方、類似構造が存在しない場合には、より手間のかかるMIR法やMAD/SAD法を用いて解析しなくてはならない。
上述の3種の方法のうち、ここでは、分子置換法(MR法)を行うための標準的なプロトコールを述べる。分子置換法は、1)非対称単位中のタンパク質分子数の見積もり、2)自己回転関数による非結晶学的回転軸の確認、3)類似タンパク質(モデル分子)の選択と編集、4)分子の回転/並進、5)結晶中の分子のパッキングの確認という5段階に分けられる。これらは、CCP4 software suite(1)がインストールされているコンピュータさえあれば、1日で行うことができる。もちろん、類似蛋白質とのアミノ酸相同性が低い場合には4)の段階で容易に結果が得られず、より多くの時間がかかることをご承知頂きたい。筆者の所属研究室では、このプロトコールにより、Phosphoenolpyruvate carboxylase (2, 3), Rubisco (4), Prostaglandin D synthase (5), Thioredoxin peroxidase(6), Thermoalkalophilic lipase (7), 5-Methylthioribose 1-phosphate isomerase (8)の構造解析に成功している。
装置
CCP4 software suite ver.6がインストールされているコンピュータ
実験手順
1)非対称単位内のタンパク質分子数の見積もり
2)自己回転関数による非結晶学的回転軸の確認
3)類似タンパク質(モデル分子)の選択と編集
4)分子の回転/並進
5)結晶中の分子のパッキングの確認
本プロトコールを始める前に
目的タンパク質のアミノ酸配列が既知であり、X線回折強度データファイル(mtz形式)が得られているということを前提として分子置換法のプロトコールの説明を始めることとする。
分子置換法の詳細
1)非対称単位内の分子数の見積もり
非対称単位とは、結晶中で行う対称操作により重ねることができない最小の領域のことを指す(詳しくは他の参考図書を参照して頂きたい)。分子置換法とは、この非対称単位にモデル分子をはめ込む操作であるので、まず、非対称単位に何分子が含まれているかを見積もることから始める。
①CCP4 InterfaceのCoordinate UtilitiesからCell Content Analysisを選択する。
②Matthews - Cell Content Analysisのパネルにて、MTZ fileとMolecular weight of proteinを入力する。空間群、格子定数、分解能が自動で表示される。
③Run Nowをクリックする。
以下のような結果が表示される。
Nmol/asym Matthews Coeff %solvent P(2.20) P(tot) 1 27.94 95.60 0.00 0.00 (略) 7 3.99 69.21 0.01 0.01 8 3.49 64.81 0.03 0.03 9 3.10 60.41 0.06 0.07 10 2.79 56.01 0.13 0.13 11 2.54 51.61 0.20 0.19 12 2.33 47.21 0.24 0.23 13 2.15 42.81 0.20 0.19 14 2.00 38.41 0.10 0.10 15 1.86 34.01 0.03 0.03 16 1.75 29.61 0.00 0.00 17 1.64 25.21 0.00 0.00 18 1.55 20.81 0.00 0.00 (略)
通常、表のMatthews Coeffで示されているVMは1.7~3.5A3/Da程度であるため(9)、この場合は、非対称単位中に8~16分子存在すると考えられる。このように、分子の数を厳密に決定する必要はなく、ある程度の可能性を検討できれば、次の段階に進む。
2)自己回転関数による非結晶学的回転軸の確認
非対称単位に複数の分子が存在することがわかれば、次に自己回転関数を計算し、非結晶学的回転軸(NCS)の存在を確認する。
①CCP4 InterfaceのMolecular ReplacementからMolrep - auto MRを選択する。 ②Molrep - Molecular Replacementのパネルにて、Self rotation functionとMTZ fileを入力する。 ③Run Nowをクリックする。
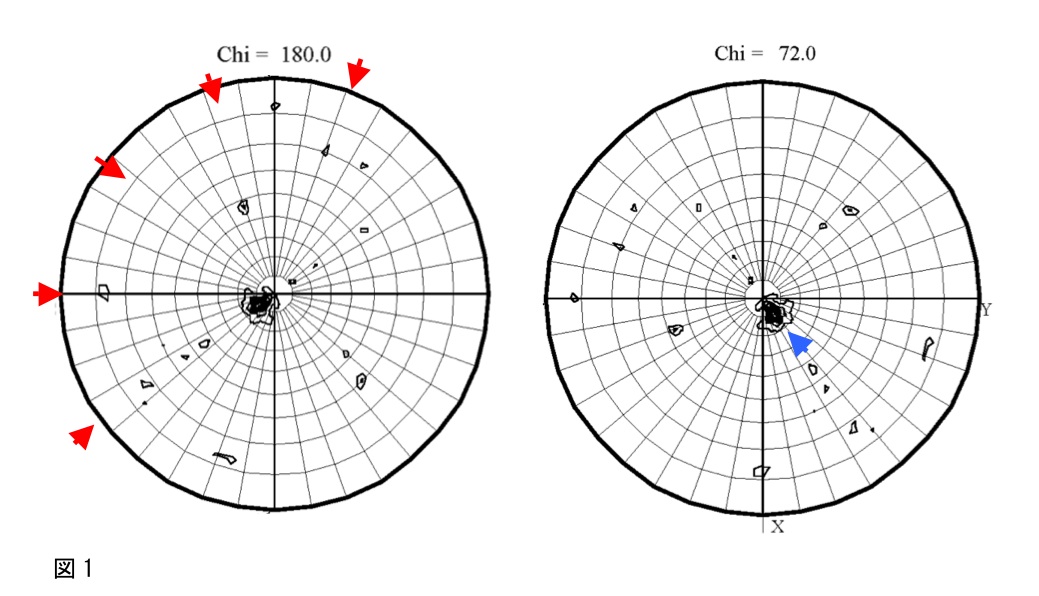
出力されたPostscriptファイルをGimpなどで開くと、極座標(polar angle; Phi, Psi)で示された図(図1)が表示される。
図1では、2回軸の存在を示すChi =180のセクションに5つの高いピーク(図1の赤矢印)が等間隔で存在しており、Chi=72°のセクションに1つの高いピーク(図1の青矢印)が存在していることから、2量体が5つ集まった10量体が存在していることが予想される。このSelf-rotation functionの計算結果と、1)で見積もった非対称単位内の分子数とを比較し、結晶中での分子のパッキングを予想しておく。
3)類似タンパク質(モデル分子)の選択と編集
まず、Protein Data Bank(http://www.pdbj.org/index_j.html)に登録されているものの中で、目的タンパク質とアミノ酸の相同性の高いタンパク質(モデル分子)を選択する。モデル分子の選択は様々な方法があるが、筆者は、Fugue(http://tardis.nibio.go.jp/fugue/prfsearch.html)やFASTA(http://fasta.ddbj.nig.ac.jp/top-j.html)といったオンラインプログラムを使用している。
①これらのWeb siteにアクセスし、アミノ酸シークエンスを入力する。相同性の高いタンパク質のPDB IDが表示される。
②Protein Data Bank(http://www.pdbj.org/index_j.html)のPDB IDに4桁のIDを入力し、Site Searchをクリックする。
③Download FilesからPDB textを選択し、ファイルをディスクに保存する。
登録されているPDBファイルには、タンパク質の構造情報が記されているATOMレコードとタンパク質名などが記されているヘッダレコードがある。また、ATOMレコードにはタンパク質の構造以外にタンパク質に結合している水分子や基質アナログなどの情報が含まれている。そのため、タンパク質の構造以外の行を削除する。加えて、複数個の分子の情報が含まれている場合もある。分子置換法には、そのうちの一分子の情報を用いるため(注複数分子用いることもある、工夫とコツの頁参照)、残りの分子の座標も削除する。例えば、下の赤字の行は、削除すべき行である(ENDは残す)。
TITLE CRYSTAL STRUCTURE OF C4-FORM PHOSPHOENOLPYRUVATE TITLE 2 CARBOXYLASE FROM MAIZE CRYST1 158.440 174.610 254.280 90.00 90.00 90.00 C 2 2 21 16 SCALE1 0.006312 0.000000 0.000000 0.00000 SCALE2 0.000000 0.005727 0.000000 0.00000 SCALE3 0.000000 0.000000 0.003933 0.00000 ATOM 1 N ILE A 35 48.657 94.067 63.704 1.00104.26 N ATOM 2 CA ILE A 35 49.991 94.402 63.186 1.00104.49 C ATOM 3 C ILE A 35 50.320 95.842 63.582 1.00104.97 C ATOM 4 O ILE A 35 51.476 96.259 63.530 1.00104.54 O ATOM 5 CB ILE A 35 50.021 94.230 61.666 1.00103.87 C ATOM 6 CG1 ILE A 35 51.421 94.535 61.167 1.00103.00 C ATOM 7 CG2 ILE A 35 48.950 95.107 61.007 1.00103.58 C ATOM 8 CD1 ILE A 35 51.639 94.176 59.752 1.00101.85 C ATOM 9 N GLU A 36 49.300 96.582 64.017 1.00104.85 N ATOM 10 CA GLU A 36 49.467 97.977 64.443 1.00103.93 C ATOM 11 C GLU A 36 49.923 98.066 65.897 1.00101.66 C (略) ATOM 7214 N ILE B 35 15.242 116.038 72.202 1.00118.93 N ATOM 7215 CA ILE B 35 13.883 115.464 71.984 1.00119.04 C ATOM 7216 C ILE B 35 13.961 113.973 71.607 1.00120.30 C ATOM 7217 O ILE B 35 12.932 113.312 71.433 1.00121.05 O ATOM 7218 CB ILE B 35 13.131 116.259 70.881 1.00117.38 C ATOM 7219 CG1 ILE B 35 11.723 115.693 70.688 1.00115.93 C ATOM 7220 CG2 ILE B 35 13.927 116.230 69.589 1.00117.17 C ATOM 7221 CD1 ILE B 35 10.904 116.419 69.659 1.00114.85 C ATOM 7222 N GLU B 36 15.184 113.446 71.511 1.00120.76 N ATOM 7223 CA GLU B 36 15.426 112.038 71.151 1.00120.12 C ATOM 7224 C GLU B 36 14.911 111.033 72.197 1.00117.94 C (略) TER 14425 GLY B 970 END
4)分子の回転/並進
ここでは、3)で準備したモデル分子を回転/並進し、結晶の非対称単位中にモデル分子をはめ込んでいく操作を行う。非対称単位中の分子数が判っていれば、その分子の個数を指定することも可能である。
①CCP4 InterfaceのMolecular ReplacementからMolrep - auto MRを選択する。
②Molrep - Molecular Replacementのパネルにて、molecular replacementとMTZ file、ならびに、Model inに準備したモデル分子のpdb fileを入力する。
③非対称単位中の分子数が判っていれば、Search ParametersのSearch for に非対称単位中の分子数(判っていれば)を入力する。
④Run Nowをクリックする。
⑤Jobが終わった後、View Files from Job-View Log File を選択して、ログファイルを確認する。以下のような結果が示される(以下、抜粋)。
回転の解が、Rf/sigmaの高い順に表示される。
theta phi chi alpha beta gamma Rf Rf/sigma
Sol_RF 1 48.01 70.44 141.80 144.68 -136.90 171.99 2496. 7.55
Sol_RF 2 71.99 109.56 321.80 78.40 145.09 113.01 2496. 7.55
Sol_RF 3 14.21 109.28 321.56 98.47 116.32 111.08 2334. 7.06
Sol_RF 4 27.68 90.71 332.91 89.71 117.38 90.71 811.5 2.45
Sol_RF 5 92.31 89.29 152.91 130.46 -120.30 134.90 811.5 2.45
Sol_RF 6 73.05 91.58 335.74 68.11 138.65 101.16 793.4 2.40
次に、回転/並進の解がCorrelation の高い順に表示される。
alpha beta gamma Xfrac Yfrac Zfrac Dens/sig R-fac Corr
Sol_TF_1 1 48.01 70.44 141.80 0.381 0.497 0.278 23.95 0.594 0.475
Sol_TF_1 2 48.01 70.44 141.80 0.377 0.499 0.218 22.47 0.613 0.429
Sol_TF_1 3 48.01 70.44 141.80 0.726 0.178 0.250 21.10 0.623 0.404
Sol_TF_1 4 48.01 70.44 141.80 0.725 0.179 0.316 20.15 0.625 0.410
Sol_TF_1 5 48.01 70.44 141.80 0.457 0.497 0.281 19.12 0.612 0.413
Sol_TF_1 6 48.01 70.44 141.80 0.396 0.509 0.334 13.36 0.629 0.414
Sol_TF_1 7 48.01 70.44 141.80 0.681 0.974 0.219 13.32 0.631 0.388
Sol_TF_1 8 48.01 70.44 141.80 0.714 0.163 0.196 13.29 0.623 0.390
Sol_TF_1 9 48.01 70.44 141.80 0.724 0.179 0.180 13.15 0.632 0.387
Sol_TF_1 10 48.01 70.44 141.80 0.788 0.165 0.262 11.97 0.616 0.385
ログファイルの最後に、Correlationの高い回転/並進の組み合わせが以下のように表示される。
Sol_Mon_1_Rf_1_Tf_1 48.01 70.44 141.80 0.381 0.497 0.278 0.594 0.475
一般に正しい解は、R-factorとCorrelationが他の解に比べて有意に高いものである。モデル分子との相同性にもよるが、50%台のR-factor になることが多い。
5)結晶中の分子のパッキングの確認
4)で得られた解の妥当性を評価するために、結晶中の分子のパッキングを確認する。
①Cootを起動させる。
②File - Open Coordinates…から4)で得られたpdb fileを選択する。座標が表示される。
③Display Managerを選択し、右のカラムのBonds (Colour by Atom)をC-alphasに変更する。
④Draw - Cell & Symmetry…を選択し、Master Switch: Show Symmetry Atoms?のYesを選択する。Symmetry by Molecule…を選択し、Display as CAsを選ぶ。続いて、Radiusを50程度に設定し、Applyをクリックすると、分子の周りに、対称操作で関係づけられた分子が表示される。
結晶中では、分子同士が相互作用していなくてはならない。したがって、上述の操作で隣接する分子を表示させた際に、分子同士が接触しているかどうかを確認する。また、分子同士が重なる場合には、解が妥当でない可能性がある。そうした場合には、分解能や分子の数を再設定し、4)の操作をもう一度行うことをお薦めする。
以上、1)~5)の分子置換法が順調に終了すれば、Refinement-Run Refmac5-rigid body refinementを行い、続いて、restraint refinementを用いて構造の精密化を行う。
工夫とコツ - 分子置換法で正しい解を得るために
分解能
分子置換法を行うにあたり、幾通りかの分解能範囲のデータを用いると良い。例えば、2.0Å分解能の回折強度データが得られている場合、筆者は、50-2.0Å, 20-3.0Å, 10.0-4.0Å, 8-6Åのように様々な組み合わせの分解能範囲にて分子置換法を行う。また、そうした場合に、ある分解能範囲のデータで分子置換した時のみ、良い結果が得られることもあるので試して頂きたい。ただし、分子置換法で解を得た後のRigid body refinementや構造精密化では、全てのデータ(例えば、50-2.0Å)を用いるのが妥当であろう。
モデル分子
筆者は、図2に示すようなD2対称(中心に222対称)を有する4量体の構造解析を分子置換法で行った際、単量体(青色の分子)、2量体(赤枠、青枠で示した2通り)、4量体全体をそれぞれモデル分子として用いて、分子置換法を試した。この中で正しい解を得たものは、唯一、赤枠で示した2量体のみであり、他のモデル分子では解が得られなかった。このように、多量体を形成しているタンパク質については、複数のモデル分子の可能性を試すことをお薦めする。
また、簡単に解が得られない場合は、ポリアラニンモデルをモデル分子として用いてもよい。(Molrepにて、The Modelから Apply convert to polyalanine & shift to originを選択する)。また、目的タンパク質とモデル分子とのアミノ酸アライメントを作成することもお薦めする。全て自動処理に任せるのではなく、自らシークエンスの相同性などを確認しておくことによって、解が得られない場合にも、相同性の低い部分を削除したモデル分子として用いるといったマニュアルによる対応が可能となる。
マニュアルフィッティング
分子置換法では、一般に、非対称単位にはめ込む分子の数が多いほど、正しい解を得ることは困難となり、ソフトウェアで全ての分子を見つけることができない場合もある。そうした場合には、一部をマニュアルではめ込むことを検討すると良い。例えば、図3のように、ソフトウェアで8分子中7分子が見つかったが、8分子目の解がどうしても見つからなかったケースがあった。ここで、分子のパッキングを確認したところ、もう一つの分子が存在すると予想される空間が存在した(図3の赤矢印参照)。このとき、タンパク質は2量体構造をとっていたために、青色の分子と2量体を形成するように1つの分子をマニュアルではめ込んで、構造解析できた。
回折強度データ
アミノ酸の相同性が高い(例えば50%以上)にもかかわらず、有意な解が得られない場合は、回折強度データの処理が正しく行われているかを再度確認して頂きたい。回折強度データ処理では、時として空間群が決定できない場合があるが、そうした場合には、無理に一つの空間群に決定する必要はない。全ての空間群でそれぞれ処理し、それぞれの回折強度データを用いて分子置換を行うと良い。
また、回折強度データそのものの質をチェックすることも重要である。データ全体の統計値(RmergeやCompleteness、Redundancy)や高角範囲の統計値のみならず、低角範囲のRmergeもチェックすると良い。これらの統計値が悪い場合には、X線照射による損傷がX線回折強度データに悪影響を与えている可能性がある。その際、フレーム毎のRmergeを確認すれば、X線照射による結晶の損傷を概ね調べることができ、Rmergeが悪いフレームは、Scalingから除くことも検討する。このように、できるだけ精度の高い回折強度データを用いて分子置換法に用いるべきである。
文献
- Acta Crystallogr D Biol Crystallogr 50, 760-3 (1994)
- Matsumura H, et al., Structure, 10, 1721-30 (2002)
- Matsumura H, et al., FEBS Lett, 458, 93-6 (1999)
- Mizohata E, et al., J Mol Biol, 316, 679-91 (2002)
- Inoue T, et al., Nat Struct Biol 10, 291-6(2003)
- Nakamura T, et al., Proteins, 62, 822-6 (2006)
- Matsumura H, et al., Proteins, 70, 592-8 (2008)
- Tamura H, Set al., Protein Sci, 17, 126-35 (2008)
- Matthews BW.. J Mol Biol, 33, 491-7 (1968)
-
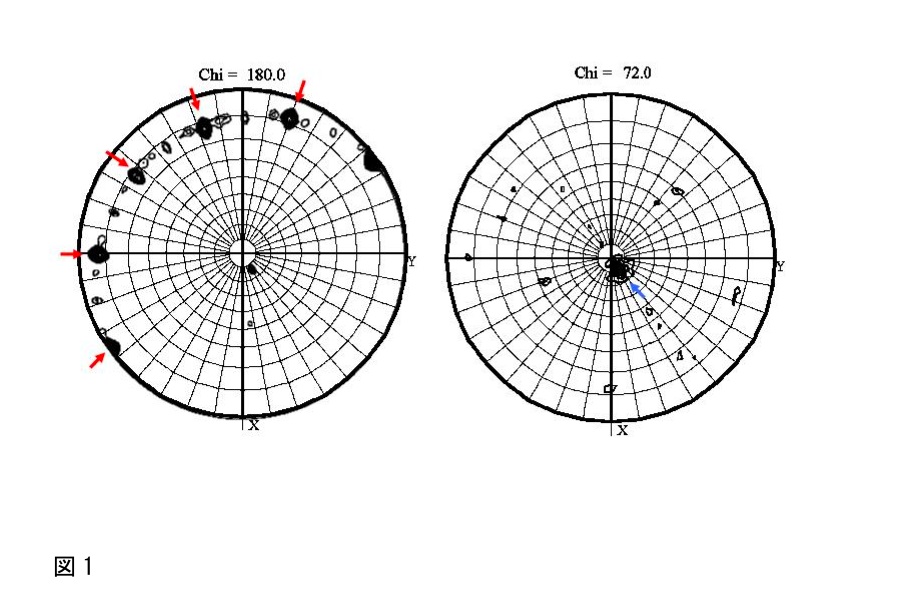
図1: -
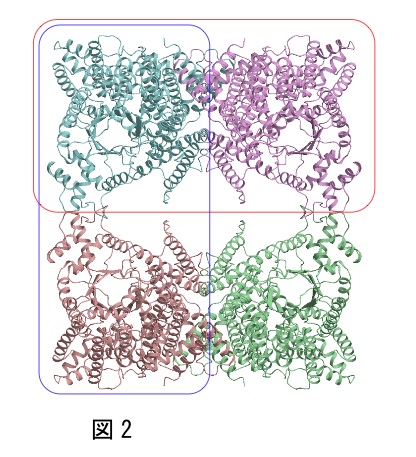
図2: -
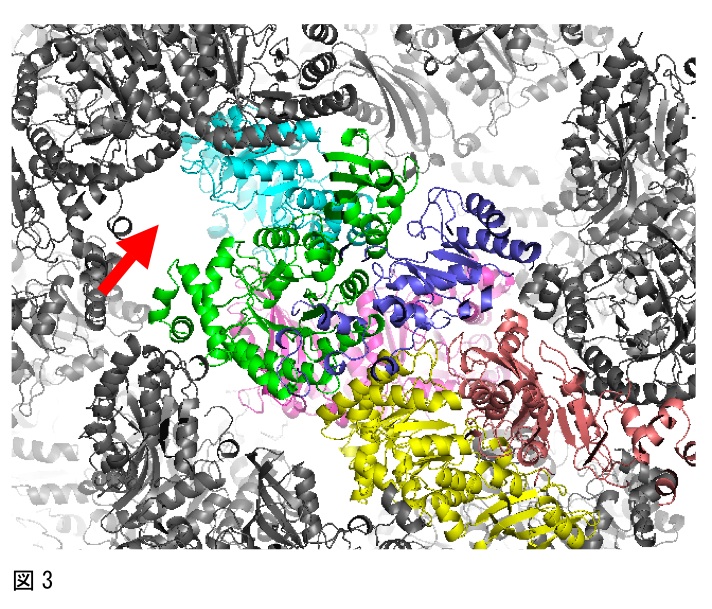
図3:

{kind=link}