

概要
Gタンパク質共役型受容体(GPCR)は細胞膜上に局在する膜タンパク質であり、生体内の多様なリガンドを受容することで様々な生命現象の制御に関与する。そのため、創薬における最大の標的の一つであり、その重要性は既承認薬の約35%がGPCRを標的としているほどである。近年、クライオ電子顕微鏡法に関する技術の革新によってGPCRの構造解析は飛躍的に進歩し、X線結晶構造解析では特に困難であったGタンパク質との複合体の構造解析の成功例が次々と報告されている。そこで本稿では、クライオ電子顕微鏡法を用いたGPCRの構造解析において頻繁に直面する問題点と、それに対処するための最新の手法について概説する。具体的には、①解離しやすいGPCR–Gタンパク質複合体に対するサンプル調製法、②柔軟な複合体に適したサンプル調製およびデータ解析の手法、③不活性状態での構造解析を可能にするサンプル調製法の3つに焦点を当てて紹介する。本稿で紹介する技術的な工夫の原理や考え方が、GPCRに限らず、広くタンパク質の構造解析において有益な知見となれば幸いである。
はじめに
Gタンパク質共役型受容体(GPCR)は、7本の膜貫通ヘリックスをもつ膜タンパク質であり、ヒト遺伝子の約1%を占める最大のスーパーファミリーを形成している。GPCRファミリーは、生体アミンやヌクレオチド、脂質などの分子をはじめ、光やタンパク質など様々な刺激を受容する(図1)。刺激となるリガンドを受容したGPCRは構造変化を起こし、細胞内でシグナリングパートナーと結合する。主なパートナーとして、Gs、Gi、Gq、G12/13の4種に大別される三量体Gタンパク質と、βアレスチンと呼ばれる多機能アダプタータンパク質が存在し、それぞれが異なるシグナル伝達経路を活性化する。特に三量体Gタンパク質は、GPCRと結合することでGDP結合状態からGTP結合状態へと変化し、GαサブユニットとGβγサブユニットに解離して下流にシグナルを伝達する。既承認薬の約35%がGPCRを標的としていることからもわかるように、GPCRは創薬標的として非常に重要であり構造研究の主要な対象であり続けている1。
近年、クライオ電子顕微鏡法による構造解析技術の発展によりGPCRの構造の報告件数は急増し、その研究は大きく進展している。クライオ電子顕微鏡法による構造解析では、画像処理の過程でアラインメント(=各粒子画像が元の3次元構造のどの角度および位置からの投影であるかを推定する過程)が行われる。この際、分子内の特徴的な領域が大きいほど、投影像の配向を判別しやすくなり、3次元再構成が容易になる。こうした手法上の特性から、主にGPCR単体を対象としてきたX線結晶構造解析とは異なり、クライオ電子顕微鏡法による構造解析ではGPCRとGタンパク質との複合体が対象とされることがほとんどである。クライオ電子顕微鏡法はGPCRの構造研究に多大な成果をもたらしたものの、そこにはいくつかの課題点が存在する。具体的には、
- 解離しやすいGPCR–Gタンパク質複合体の構造解析では、サンプル中の複合体粒子数が極端に少なくなるため、構造決定に至らない場合が多い。
- 複合体の柔軟性によって、密度マップの質が大きく低下する。
- 不活性状態の構造解析は、Gタンパク質との複合体を形成しないため難しい。
の3点が挙げられる。そこで本稿では、これら3つの課題を克服すべくこれまでに確立されてきた手法を解説する。また、これらの手法や考え方は、GPCRに限定されるものではなく、他のタンパク質の構造解析にも応用可能であることを併せて述べる。
1. 解離しやすい複合体のためのサンプル調製法
クライオ電子顕微鏡法では、撮影したマイクログラフ上の多数の粒子から目的の粒子を選別・平均化することで、高分解能の密度マップを再構成する。解離しやすいGPCR–Gタンパク質複合体は、ゲルろ過カラムクロマトグラフィーなどによって複合体を分画したとしても、グリッド作製時に解離してしまうことがある。このような場合、目的の複合体の選別・平均化に必要なマイクログラフの数、そしてデータ解析の計算量が大幅に増加し、構造決定に至らない場合が多い。
GPCRに結合したGタンパク質は、GDP/GTP交換反応ののち、GαサブユニットとGβγサブユニットに解離する。この際、最も構造的に安定したGPCR–Gタンパク質複合体の状態は、ヌクレオチド非結合状態である。そのため、構造解析にあたっては、GDP/GTP結合部位を欠失させ、かつ熱安定化変異を導入したminiGタンパク質(miniGs,miniGo,miniG12等)が頻繁に利用されてきた(図2)2。ただし、Giについては性状の良いminiGタンパク質が確立されていない。そのため、Giと共役するGPCRの構造解析では、精製中にジホスホヒドロラーゼであるApyraseを添加し、サンプル中のGTPやGDPをGMPへと加水分解することで、Gi三量体の解離を防ぐ手法が用いられる。また、GsやGiに結合するGPCRの構造解析ではNanobody 35(Nb35)とsingle-chain variable fragment 16(scFv16)と呼ばれる構造認識抗体がよく用いられる3,4。これらの抗体には三量体Gタンパク質の解離を抑制する効果があり、特にscFv16はGαのN末端とGβγとの界面を認識するため、GqやG12/13のN末端数十残基をGiのものに置換することで、それらと共役するGPCRの構造解析にも利用可能である5,6。さらに、GαのN末端にGγを十分な長さのリンカーを介して融合することで、三量体Gタンパク質の解離を防ぐ手法も用いられる7。
最近では、スプリットルシフェラーゼの各断片であるLgBiTとHiBiTを、それぞれGPCRとGβのC末端に融合する手法が利用されている(図3)8。両者は非常に高い親和性をもち(解離定数:約700 pM)、GPCRとGタンパク質との安定した複合体の形成を促進する。また、両者の間には十分な長さのリンカーが挿入されており、スプリットルシフェラーゼの融合が複合体構造に影響を及ぼした例は現在のところ確認されていない。
また、非常に難易度が高いGPCR–βアレスチン複合体の構造解析では、GPCRのC末端をバソプレシン2型受容体のものに置換する戦略が取られる。βアレスチンはリン酸化修飾を受けたGPCRのC末端を認識し結合するが、数あるGPCRの中でもバソプレシン2型受容体のC末端はβアレスチンに対して高い親和性をもつ。これにより安定な複合体形成が可能となり、構造解析が現実的な難易度まで下がるため、本手法はこれまでに報告された多くのGPCR–βアレスチン複合体の構造研究で使用されてきた9–12。
2. 柔軟な複合体のためのサンプル調製・データ解析法
クライオ電子顕微鏡法によるGPCR–Gタンパク質複合体の構造解析では、サイズが大きく構造的な特徴をもつGタンパク質部分がアラインメントの中心となることが多いため、GPCR部分の局所分解能はGタンパク質部分に比べて低くなる傾向がある。また、GPCR–Gα界面とGα–Gβ界面にも高い柔軟性が存在しており、これはGPCR部分の局所分解能をさらに低下させる要因となっている(図4)。これらの柔軟性に起因する構造の不均一性(heterogeneity)は、分解能の異方性、すなわち特定の方向から見た分解能と別の方向から見た分解能が異なる現象を引き起こす。なお、これは特定の角度の投影が存在しないこと(オリエンテーションバイアス)による異方性とは異なる原因によるものであり、明確に区別されるべきである。こうした局所分解能の悪化や分解能の異方性といった問題を克服するため、その根底にある複合体の柔軟性に対処する取り組みが進められてきた。
その一つが抗体による界面の安定化である。先述したNb35はラマ由来の抗体であり、GαsとGβの界面を固定することで柔軟性を大幅に抑制する効果がある(図5)。初めて報告されたGPCR–Gタンパク質複合体構造は、X線結晶構造解析およびクライオ電子顕微鏡法のいずれによるものもGPCR–Gs–Nb35複合体の構造であったことから、構造研究におけるNb35の有用性が伺える3,13。こうした有用性から、Nb35をGs共役型GPCR以外にも適用する手法の開発が進められてきた。これまでに、視覚ロドプシン–Gt(トランスデューシンとしても知られる)複合体の構造研究において、GαtとNb35をそれぞれ改変して結合するようにしたeGαtとNb35*の組み合わせが開発されている14。さらに筆者らは、GαtとGαiが高い配列相同性をもつ点に着目し、この方法がGi共役型GPCRの構造解析にも適用可能であることを示した15。
また、データ解析の工夫によっても、複合体の柔軟性をある程度克服することが可能である。特に有用な手法として、GPCR部分にフォーカスして3次元クラス分類を行う方法が挙げられる(図6)。具体的には、まず2次元および3次元クラス分類により、解離した粒子や氷の像を除去した後に3次元再構成を行う。得られた密度マップをもとに、ChimeraXなどのソフトウェアを用いることでGPCR部分のみを覆うマスクを作成する。その後、このマスク領域に限定して、再度アラインメントを実施することなく構造的特徴に基づいた3次元クラス分類を行う。アラインメントなしのクラス分類を行うことで、角度や位置情報を維持しつつ、GPCRとGタンパク質の相対位置が均一な粒子を選別することができる。最後に、同様のマスクを用いてGPCR部分に限定し、アラインメントの精密化を行うことで、柔軟性の影響を抑えた密度マップを得ることが可能となる。一連の作業の際、RELIONやCryoSPARC、あるいは3D-FSCウェブサーバー(http://3dfsc.salk.edu/upload/)などの機能を活用して、局所分解能や分解能の異方性を可視化できる16–18。特に、CryoSPARCや3D-FSCウェブサーバーを用いたDirectional Fourier Shell Correlation(Directional FSC)のプロットは、分解能の異方性を評価する上で非常に有用である。この指標は、標準的なFSCを方向別に計算することで得られる。これらを適宜確認し、問題点を整理することが密度マップの質の向上につながるだろう。
クライオ電子顕微鏡法において広く使用されている解析ソフトウェアとして、RELIONやCryoSPARCが挙げられる。RELIONは、MRC分子生物学研究所のグループによって開発された無料のオープンソースソフトウェアであり、クライオ電子顕微鏡法の黎明期から長期にわたりデータ解析に利用されてきた。一方、近年では、Structura Biotechnology Inc. が開発したCryoSPARCが広く使用されるようになっている。CryoSPARCは、非営利の学術利用に限り無料で使用でき、公式ドキュメントが充実している。また、CryoSPARC discuss(https://discuss.cryosparc.com/)を参照することで、実用的なパラメーター設定や頻発するトラブルへの対処が容易である。
3. 不活性状態での構造解析に向けたサンプル調製法
GPCRを標的とする薬剤の多くはGPCRの活性を抑制する拮抗薬や逆作動薬である。しかし、これらの薬剤はGPCRを活性化しないため、薬剤結合状態のGPCR–Gタンパク質複合体を調製することは事実上不可能である。一方で、GPCRのみでは大きさや構造的な特徴が不十分であるため、現状、クライオ電子顕微鏡法によって不活性状態GPCRのみを構造解析することは非常に困難である。そこで、GPCRを他の安定なタンパク質と融合させ、粒子の大きさと特徴を増加させるフィデューシャルマーカーとして利用することで構造解析を可能にする手法が開発されてきた。
細胞内ループ3にアポ型シトクロムb562変異体(BRIL; ~11 kDa)を挿入する手法は、もともとX線結晶構造解析において、結晶内でのパッキングを向上させる目的で用いられてきた19。こうした背景から、BRILの融合はクライオ電子顕微鏡による構造解析においても早期に注目された手法である。具体的には、細胞内ループ3にBRILを融合し、さらに抗BRIL抗体のFabフラグメント(~50 kDa)を添加して大きさと構造的な特徴を大幅に増加させる手法である(図7)。この手法により、アデノシンA2A受容体、Frizzled 5受容体の不活性構造が3.4–3.7 Åの分解能で報告されている20,21。
また、カルシウム/カルモジュリン依存性セリン/スレオニンフォスファターゼであるカルシニューリン(~59 kDa)を融合する手法も報告されている(図8)。カルシニューリンは、CN-AとCN-Bの2つのサブユニットで構成され、低分子化合物FK506依存的にFKBP12(~12 kDa)という小さなタンパク質によって阻害される。そこでGPCRのC末端にCN-Aを、細胞内ループ3にCN-Bをそれぞれ融合し、FKBP12とFK506を添加することで、安定して結合したGPCR–カルシニューリン複合体を得ることができる。この手法を用いて、現在までにβ2アドレナリン受容体、エンドセリンB型受容体の不活性構造が3.3–3.5Åの分解能で報告されている22,23。
さらに、Nanobody 6(Nb6; ~12 kDa)と呼ばれる抗体を用いた手法も報告されている(図9)。Nb6は不活性状態のκオピオイド受容体の膜貫通ヘリックス5と6を認識する抗体である。そこで、Nb6結合部位に対応する残基をκオピオイド受容体のものに置換することで、他のGPCRにもNb6が結合するようになり、構造決定が可能になる。また、Nb6の改変型抗体であるMb6やNb6Mを使用することで、さらに構造解析が容易になる。Mb6は認識部位を保持しつつ、大きさと構造的な特徴を増加させた改変体(~56 kDa)であり、Nb6Mはそれ自体に対してさらに別のFab(~49 kDa)を結合可能にした改変体である。この手法を用いて、これまでにμオピオイド受容体、ソマトスタチン2型受容体、ニューロテンシン1型受容体、ヒスタミン2型受容体の不活性構造が報告されている24。これらの構造は2.4–3.1 Åと分解能が高く、水分子や陽イオンを含めた議論が可能となっている。
以上3つのいずれの手法においても、GPCR部分とマーカー部分(BRIL、カルシニューリン、抗体)の相対位置は、構造の不均一性を抑えるためなるべく一定にする必要がある。これを満たすような剛直なコンストラクトを設計するために、AlphaFoldによる構造予測や、それを用いた分子動力学シミュレーションが有用であることが報告されている25。また、一般に剛直なタンパク質は熱安定性が高いことを考慮し、蛍光ゲルろ過クロマトグラフィー法を用いた熱安定性評価を行うこともコンストラクトの検討に有効だろう26。
おわりに
本稿では、近年急速に発展を続けているクライオ電子顕微鏡を用いたGPCRの構造解析 において、頻繁に直面する3つの問題点と、その対処法について概説した。なお、本稿で紹介した手法はGPCR以外のタンパク質の構造解析でも活用できるものである。例えば、不安定で解離しやすい複合体の構造解析では、リンカーやスプリットプロテインを用いることで精製時の収量や測定データ中の複合体割合が増加する可能性がある。また、目的タンパク質の柔軟性が密度マップへ悪影響を及ぼす現象はGPCRに限らず広くみられる。このような場合、特定の部分にフォーカスしてアラインメントなしで行う3次元クラス分類は、一般的に有効な対処法の一つである。そして、サイズが小さく構造的な特徴が少ないためにクライオ電子顕微鏡による構造解析が困難な膜タンパク質に対して、マーカーとなる別のタンパク質を融合する戦略は他の膜タンパク質でも実践されつつある27。本稿で紹介した問題点と対処法の根底にある原理や考え方の理解が、GPCRをはじめとする多くのタンパク質の構造解析の一助となれば幸いである。
文献
- Sriram, K. et al., Mol. Pharmacol., 93, 251–258 (2018).
- Nehmé, R. et al., PloS One, 12, e0175642 (2017).
- Zhang, Y. et al., Nature, 546, 248–253 (2017).
- Koehl, A. et al., Nature, 558, 547–552 (2018).
- Kim, K. et al., Cell, 182, 1574–1588.e19 (2020).
- Qian, Y. et al., Mol. Cell, 82, 4340–4352.e6 (2022).
- Sano, F. K. et al., eLife, 12, e85821 (2023).
- Akasaka, H. et al., Nat. Commun., 13, 5417 (2022).
- Lee, Y. et al., Nature, 583, 862–866 (2020).
- Staus, D. P. et al., Nature, 579, 297–302 (2020).
- Liao, Y.-Y. et al., Cell, 186, 5784–5797.e17 (2023).
- Chen, K. et al., Nature, 620, 904–910 (2023).
- Rasmussen, S. G. F. et al., Nature, 477, 549–555 (2011).
- Gao, Y. et al., Mol. Cell, 75, 781–790.e3 (2019).
- Oshima, H. S. et al., Biochem. Biophys. Res. Commun., 693, 149361 (2024).
- Punjani, A. et al., Nat. Methods, 14, 290–296 (2017).
- Kimanius, D. et al., Biochem. J., 478, 4169–4185 (2021).
- Tan, Y. Z. et al., Nat. Methods, 14, 793–796 (2017).
- Liu, W. et al., Science, 337, 232–236 (2012).
- Tsutsumi, N. et al., eLife, 9, e58464 (2020).
- Zhang, K. et al., Nat. Commun., 13, 4366 (2022).
- Xu, J. et al., Proc. Natl. Acad. Sci., 121, e2414544121 (2024).
- Shihoya, W. et al., 2023.12.30.573741 Preprint at https://doi.org/10.1101/2023.12.30.573741 (2024).
- Robertson, M. J. et al., Nat. Struct. Mol. Biol., 29, 1188–1195 (2022).
- Jumper, J. et al., Nature, 596, 583–589 (2021).
- Hattori, M. et al., Structure, 20, 1293–1299 (2012).
- Chen, H. et al., Cell, 186, 2644–2655.e16 (2023).
-
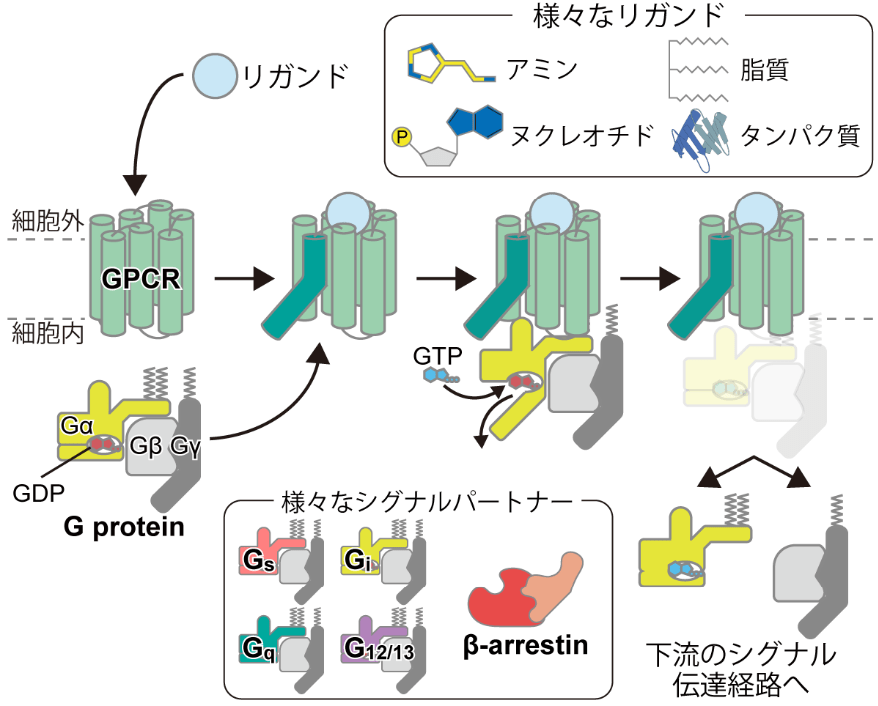
図1:GPCRによるシグナル伝達機構
各種GPCRは特異的にリガンドを認識し、特定のパートナーを介して下流へシグナルを伝達する。 -

図2:miniGタンパク質と抗体の模式図 -
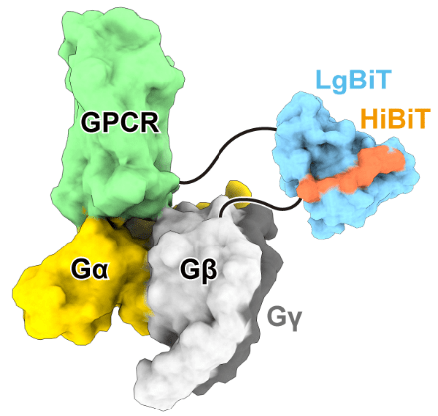
図3:スプリットルシフェラーゼによる複合体安定化の模式図 -
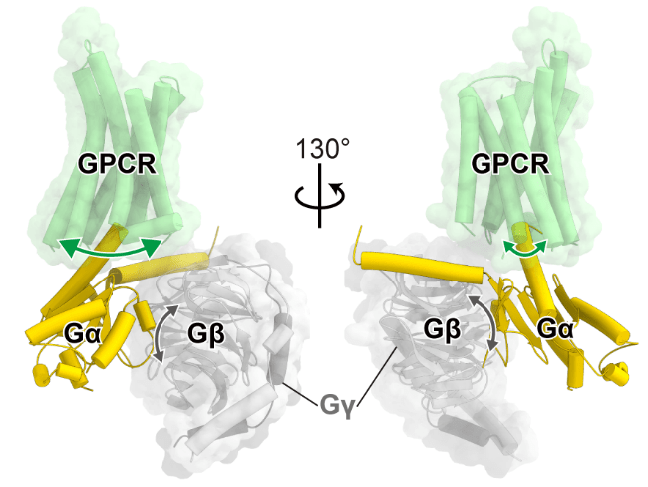
図4:GPCR–Gタンパク質複合体における柔軟な界面
矢印の大きさが柔軟性の程度を表す。 -
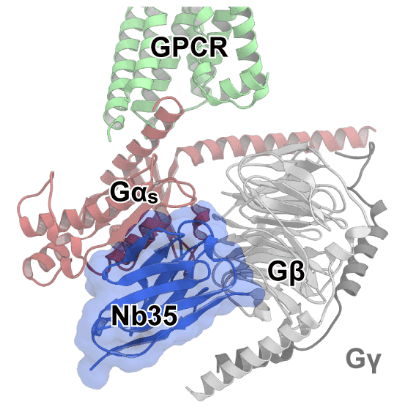
図5:Nd35の認識部位
Nd35はGαsとGβの界面を認識する。 -
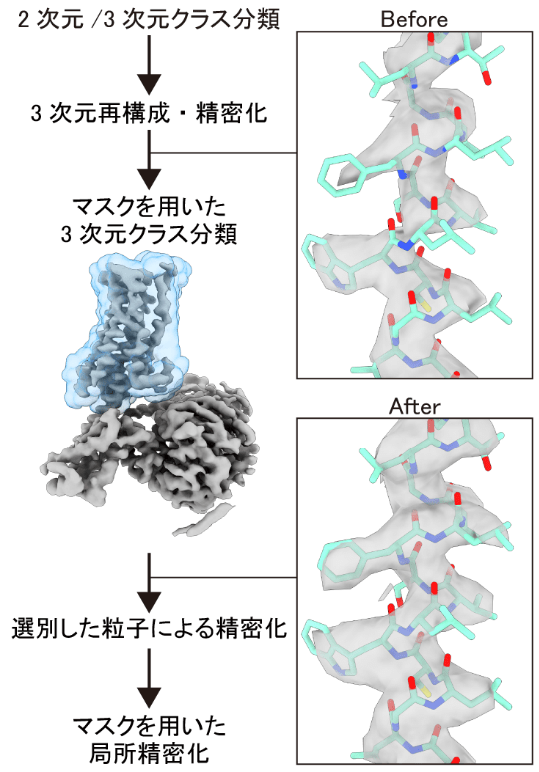
図6:クライオ電子顕微鏡によるGPCRの構造解析ワークフロー
3次元クラス分類の前後の密度マップを等しいσで示す。 -
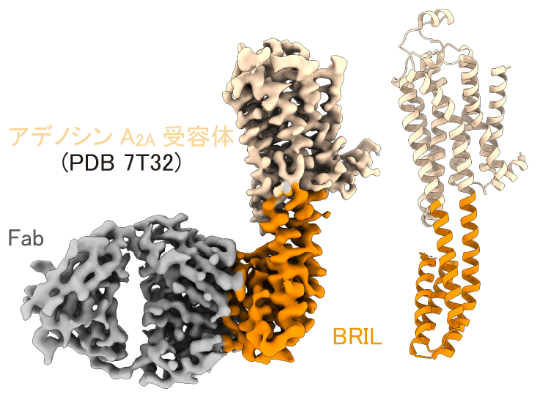
図7:BRILが融合したGPCRの不活性構造
BRILを認識するFabはモデルが構築されていないため表示していない。 -
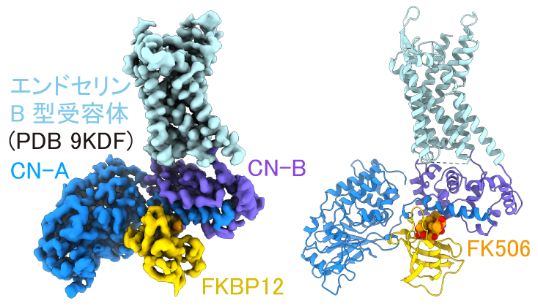
図8:カルシニューリンが融合したGPCRの不活性構造
カルシニューリンの両断片がFKBP12とFK506によって固定されている。 -

図9:Nb6が結合したGPCRの不活性構造
逆作動薬結合部位付近の密度とモデルを示す。
