

- 概要・目次
- はじめに
- hFasLECD 誘導体の *Pichia pastoris* 分泌発現生産系を用いた調製
- hFasLECD の部位特異的化学修飾体の調製とキャラクタリゼーション
- 今後の展望
- 謝辞・引用文献
概要
ヒト Fas リガンドはがんなどの重大な疾病の原因となる細胞に対して細胞死を誘導する能力を有する膜蛋白質であり、その細胞外ドメインは標的細胞上のFasレセプターに特異的に結合しアポトーシス実行のためのシグナル伝達の引き金を引く役割を担っている。この総説では、ヒト Fas リガンド細胞外ドメインを対象とした Pichia pastoris 酵母を宿主とする各種誘導体の発現生産系の構築および部位特異的化学修飾体の調製を手段とする医療応用上有用な機能を持つ蛋白質分子の創製を目指した分子デザインのための方法の開発に焦点を当てて紹介する。本総説では、関連する周辺の研究の進展ならびにこれまでに発表した総説や原著論文において充分に詳しく記述できなかった事項や一部未発表のデータも含めて、経緯と現状を省みると共に、今後の研究の発展への期待を含めた展望についても触れる。尚、本稿は著者が2018年1月に発表した英文総説[1]に加筆と修正による改訂を加えたものを基に、その後に発表した原著論文[2]の内容を追補して作成したものである。
目次
- はじめに
- 1.1 ヒト Fas リガンドとは
- 1.2 Pichia pastoris を宿主とする異種蛋白質の発現生産系
- 1.3 これまでに開発されてきたヒト Fas リガンド細胞外ドメイン(hFasLECD)の異種発現生産系
- 1.4 これまでに開発されてきた医療応用を目的とした hFasLECD 誘導体
- hFasLECD 誘導体の Pichia pastoris 分泌発現生産系を用いた調製
- 2.1 分泌発現生産系の構築
- 2.2 タグ配列付加ならびに糖鎖付加部位欠失の影響
- 2.3 N 末端領域の除去が分泌生産量に与える効果
- 2.4 陽イオン交換クロマトグラフィーによる精製
- 2.5 ヒト Fas レセプター細胞外ドメインに対する結合活性の確認
- 2.6 ディスポーザブルカルチャーバッグを用いた培養システムの開発
- hFasLECD の部位特異的化学修飾体の調製とキャラクタリゼーション
- 3.1 N 末端タグ配列中にシステイン残基を有する誘導体の設計と発現生産
- 3.2 部位特異的化学修飾体の調製法の検討
- 3.3 低分子性蛍光色素による化学修飾体の調製
- 3.4 他の機能性蛋白質分子との架橋体の調製
- 3.5 共有結合構造の同定と細胞死誘導活性の評価
- 今後の展望
- 謝辞
- 引用文献
1. はじめに
1.1 ヒト Fas リガンドとは
ヒト Fas リガンド(hFasL)は、ヒトゲノムの第1番染色体上にコードされ[3]、腫瘍壊死因子(tumor necrosis factor/TNF)レセプタースーパーファミリーと名付けられた蛋白質グループの一員であるヒト Fas レセプター(hFasR/CD95/APO-1)を表出する標的細胞に対してアポトーシスによる死の引き金をひく、一回膜貫通型の糖蛋白質である。hFasR に代表される、いわゆるデスレセプターと結合して標的細胞の細胞死を誘導する性質のある蛋白質は一般的にデスリガンドと呼ばれている。FasL(CD95L/CD178/APO-1L)は TNFα、リンフォトキシンα(TNFβ)、TRAIL(CD253/APO-2L)、TWEAK(CD255/APO-3L)などと並んで代表的なデスリガンドの一つである。これらの蛋白質群は TNF リガンドスーパーファミリーと呼ばれ、標的細胞に対するアポトーシスによる死の誘導という注目すべき生物活性のために、その発見、同定以来、がんや関節リウマチ、全身性エリテマトーデスに代表される各種の自己免疫疾患などの重大な疾病の治療への貢献を目的とした数多くの研究がこれまでになされてきた。
FasL-FasR 系によるアポトーシス実行の特徴ならびにその医療応用の可能性を対象とした優れた和文の総説や解説についてはこれまでに多数発表されてきている。それらの中で最も解りやすくかつ包括的なものの一つに、Fas 抗原の発見者の一人でありその命名者でもある米原伸博士らと共に FasR の cDNA のクローニングによる同定[4]を行われた長田重一博士が共同の編集委員を務められた日本生化学会編の「シリーズ・バイオサイエンスの新世紀 第6巻 細胞の誕生と死」の中に含まれている、FasL 遺伝子のクローニングに初めて成功された須田貴司博士著の「第3章 細胞死:②FasとFasリガンド」が挙げられる[5]。また、英文の総説集として代表的なものの一つに、Herald Wajant 博士編の “Fas Signaling”[6]がある。FasL の発見と遺伝子のクローニングに関する経緯、基本的なシグナル伝達経路ならびにアポトーシスの誘導に関わる生体内での主要な生理的役割や病理的機能についてはこれらの著書にほぼ網羅されている。さらに、FasL-FasR 系を介するシグナル伝達経路は標的細胞の状態に応じて、アポトーシスだけではなくネクロプトーシスや細胞分化の誘導などにも関与することが指摘されており[6]、近年ではそれらの面からの自己免疫疾患や悪性腫瘍に対する免疫学的治療の可能性についても積極的に検討されるようになってきた[7]。
図1に hFasL 蛋白質を構成する全体的なドメイン構造を、また図2にその中で細胞外ドメイン部分の一次構造ならびに二次構造を示した。ヒト FasL 細胞外ドメイン(hFasLECD)については、以前から生理的条件下において同一サブユニットからなるホモ3量体構造を形成し、高さ約 6 nm の頂上部が欠損したベル型のピラミッド状の立体構造を有することが予測されていたが[8]、具体的な形状については、近年まで上記の「第3章 細胞死:②FasとFasリガンド」中に掲載されているコンピュータを用いた予想に基づいて作成された「Fas-Fas リガンド複合体の立体構造モデル」などが主なよりどころであった。その後2013年になり、X 線結晶構造解析を用いた詳細な立体構造に関する情報が米国アルバートアインシュタイン医科大学の Weifeng Liu 博士を筆頭とする Steven C. Almo 博士らのグループにより、現在のところ hFasL がヒトの体内において hFasR 以外に唯一特異的に結合すると考えられているデコイ(おとり)型レセプターであるヒト DcR3(hDcR3)[9]との複合体の形で蛋白質構造データバンク(PDB)に PDB ID:4MSV(公開日:2013-11-27)として登録された[10]。しかしながら、未だ野生型 hFasLECD 単体ならびにそれに対するアポトーシスシグナル伝達実行型受容体の細胞外ドメインであるヒト FasR 細胞外ドメイン(hFasRECD)との複合体の立体構造は明らかにされていない。そのため、野生型 hFasLECD の複合体形成過程における構造変化や複合体形成時の hFasRECD の場合と hDcR3 の場合の詳細な相互作用様式の相違の解明等については今後の検討課題として残されている。一方、hFasLECD 中のアミノ酸残基番号164-169に対応する AspThrTyrGlyIleVal の配列が同じく TNF リガンドスーパーファミリーに属する蛋白質分子の一つである TL1A 中の対応する配列である HisGluLeuGlyLeuAla に置換された変異体についての単体ならびにその hDcR3 との複合体については既に PDB に登録されている(PDB ID:5L19および5L36[10])(図3)。X 線結晶解析で明らかにされた立体構造によると、後述するアミノ酸残基番号139-281の領域は主に逆並行型のβ-シート構造からなる二次構造が発達しており(図2)、多くの水素結合を介した相互作用に裏打ちされたゆらぎの少ない熱力学的に安定な構造であることが示唆される。この二次構造形成領域は2002年に TNF リガンドスーパーファミリーに属する他の蛋白質との配列相同性から予測されたもの[8]と、よく一致していた。
hFasL 蛋白質分子を構成する各領域のうち、細胞外ドメインは標的細胞の表面に存在する hFasR に特異的に結合することでアポトーシス実行のためのシグナル伝達の引き金を引く役割を担っている。本稿では、この細胞外ドメインに関する各種誘導体の Pichia pastoris を宿主とする分泌発現生産系の構築ならびに部位特異的化学修飾を手段とした他の機能性分子との結合体の調製法の開発に焦点を当てて記述するが、本論に入る前にまず、P. pastoris を宿主とする異種蛋白質の発現生産系、これまでに開発されてきた hFasLECD の異種発現生産系ならびに将来的な医療分野での応用を目的として開発されてきた hFasLECD 誘導体について概略的に紹介する。
1.2 Pichia pastoris を宿主とする異種蛋白質の発現生産系
メタノール資化性酵母の一種であるピキア酵母(Pichia pastoris)は、単細胞性の微生物であることから蛋白質の発現生産実験を行う上で大腸菌と同様な取扱いの簡便さを有するだけでなく、同時に真核生物でもあるため哺乳動物細胞の場合と類似した品質管理機構を伴う高度な分泌生産能力を有している。そのため、ヒト由来のものを含む異種蛋白質の発現生産において多用され、近年ではゲノム編集を始めとする各種の合成生物学的手法による宿主としての改良も盛んに行なわれている[11,12]。一般的な異種組換え蛋白質生産用宿主としての P. pastoris の優位性や特徴についてはこれまでに発表された多くの総説やモノグラフ等において触れられているので、ここでは筆者が研究を進める過程でしばしば参照し、具体的な実験を行なうに当たって大変有用であった詳細なプロトコールが記載されている成書や技術資料について紹介するにとどめる。
P. pastoris を使用した異種蛋白質の発現生産系の開発に関する代表的な英文プロトコール集としては、いずれも Methods in Molecular Biology シリーズの一つとして Humana Press より刊行されている、David R. Higgins 博士と James M Cregg 博士により編集された “Pichia protocols”(1998年)[13]ならびに、その続編の “Pichia protocols Second Edition”(2007年)[14]が挙げられる。また P. pastoris を使用した発現生産実験に関する詳細な記述がされている和文のプロトコールの例としては羊土社より刊行されている実験医学別冊「目的別で選べる蛋白質発現プロトコール」(2010年)の第3章「3. 酵母、③タンパク質の発現」の項[15]に含まれている、内藤忠相博士と杉山賢司博士により著された「2. ピキア酵母におけるメタノールを用いたタンパク質発現-小スケールでの発現チェックおよび培養条件の至適化」ならびに「3. ピキア酵母におけるメタノールを用いたタンパク質発現-大スケール」の2つの解説記事、加えて日本蛋白質科学会編のオンライン版プロトコール集である「蛋白質科学会アーカイブ」中の「蛋白質の調製」のセクションに含まれている櫻井一正博士による「#018 メタノール資化性酵母 Pichia pastoris を用いた組換え蛋白質の発現―発現系の特徴と手順」(2008年)[16]や本稿の2.6項で後述する「#078 ディスポーザブルカルチャーバッグを利用したメタノール資化性酵母 Pichia pastoris による組換え蛋白質の分泌発現生産」(2014年)[17]などがある。
1.3 これまでに開発されてきたヒト Fas リガンド細胞外ドメイン(hFasLECD)の異種発現生産系
筆者は2012年に FasL や FasR を含むいわゆるデスリガンドならびにデスレセプターの細胞外ドメイン全般に関するそれまでの異種発現生産系の開発状況に関する総説を発表した[18]。ここでは、hFasLECD に限って、その後の進展を含めて概述する。hFasL 蛋白質は1回膜貫通 II 型の蛋白質であり、その構造遺伝子領域の解析から、N 末端側のドメイン(80残基)が細胞内に局在し、C 末端側の細胞外ドメイン(179残基)との間に22アミノ酸残基からなる膜貫通領域が存在することが明らかになっている(図1)。いわゆる分泌シグナル配列は N 末端部位には存在せず、翻訳された蛋白質が C 末端側ドメインを細胞外に表出した形で膜上に局在化するためのシグナルは中央の膜貫通部位に存在していると考えられる。完全な構造を持つ生物学的に活性な膜蛋白質としての hFasL は生理的条件下で3つの同一サブユニットからなるホモ3量体として存在するが、サブユニット当りジスルフィド結合1か所を含むその細胞外ドメイン部分のみを抜き出して生産した場合にも、正常な立体構造が形成された発現産物は3量体を形成する。細胞外ドメイン部分のみを活性型3量体として生産させるに当たっては、後述するようにそのN末端に分泌シグナル配列を人為的に付加し、酵母などの真核生物の分泌発現系を利用するのが有利である。具体的な発現生産系としては、これまでに P. pastoris[19,20]ならびに細胞性粘菌の一種であるキイロタマホコリカビ(Dictyostelium discoideum)を宿主とするもの[21]などが知られており、これらの宿主を使用した場合にはサブユニット当り3か所存在する N 結合型糖鎖付加部位への翻訳後修飾が生じると考えられる。
一方、分泌生産経路があまり高度には発達していない原核生物である大腸菌(Escherichia coli)を宿主とする菌体内発現系を用いる場合には糖鎖付加は起こらず、そのままでは正常な立体構造形成が生じ難いため、通常不溶性の発現産物として蓄積される[10,22]。先に述べた hDcR3 との複合体のX線結晶解析実験においては、大腸菌の菌体内発現系[23]により得られた不溶性産物を対象として試験管内で立体構造を再構成[24,25]させることにより調製された野生型ならびに変異型の可溶性 hFasLECD の試料が、昆虫由来の培養細胞の一種であるショウジョウバエの S2 細胞を用いて生産された hDcR3[26,27]との複合体形成に用いられた。当該論文中の記述によると野生型の hFasLECD 試料単独での結晶化は困難であり、真核生物で起こる糖鎖付加は単に水溶性を高めることに寄与しているだけではなく、凝集体の形成を抑止しているようである。
1.4 これまでに開発されてきた医療応用を目的とした hFasLECD 誘導体
細胞外ドメイン部分のみからなる可溶型の hFasLECD は単独の3量体分子の場合にはたとえ標的細胞表面上の FasR に結合しても、それだけでは効率良くアポトーシスを誘導することができない。また、ある種の低分子性抗がん物質によるアポトーシスの誘導においては、細胞表面を構成する脂質二重膜上でのラフト(筏)構造の形成等を介した FasR の会合が起こり、それに引き続くアポトーシスの実行を促進すると考えられている[28]。そのため、蛋白質工学を用いた分子デザインの観点から、細胞膜上で隣接する少なくとも2分子の FasR を会合させるべく、hFasLECD に改変を加え hFasLECD-hFasR 複合体のオリゴマー形成を促進させることを目的に各種の工夫が行われてきた。中でもヒトアディポネクチンのコラーゲン様ドメインが付加された誘導体である MegaFasL[29]は2分子の3量体型 hFasLECD からなる6量体構造を形成し、ヒトの悪性脳腫瘍の原因となるグリオーマ細胞やその幹細胞様細胞に対してアポトーシスを誘導する能力を示すことが明らかにされている[30,31]。MegaFasL はハムスター卵巣由来の CHO 細胞を用いた工業的生産が可能[32]であり、製品名 APO010(Oncology Venture 社)として各種のがん細胞に対する細胞死誘導作用が確認され、再発型あるいは難治型の多発性骨髄腫患者を対象とする第 I 相および第 II 相の臨床試験(Clinical Trial.gov, ID:NCT03196947)も行われた。また、インターロイキン6ファミリーに属するサイトカインの一種である白血病阻止因子レセプター gP190 の D1、Ig および D2 などのドメインの付加によるオリゴマー形成についても検討され[33]、Ig ドメインを付加したものは6量体ならびに12量体を形成し、ヒトの類表皮腫が移植された免疫不全マウスに対して延命効果を示すことが明らかになった。
一方、hFasLECD の治療用蛋白質としての開発を検討する上で、最も重要な点の一つは全身性投与の際に生じると考えられる肝臓を始めとする hFasR を介したアポトーシス誘導に対して感受性の高い正常な臓器への重篤な副作用の軽減である[34]。この課題を克服するために、標的細胞へのターゲティング能力を付与させるための数多くの工夫が施されてきた。代表的な戦略として、標的とするがん細胞表面上に豊富に発現する抗原に特異性を有する単鎖化可変部ドメイン(scFv)型の抗体フラグメントとの遺伝子レベルでの結合による融合蛋白質の調製が挙げられる。これまでにB 細胞を起源とするリンパ腫細胞上の CD20 抗原や T 細胞を起源とする白血病細胞上の CD7 抗原などをターゲットとして、HEK293 細胞や CHO 細胞などの哺乳動物由来の培養細胞で発現生産[35,36]された融合蛋白質が試験に供され、培養細胞ならびに実験動物個体において一定の増殖抑制効果が確認されている。この手法を用いて医療応用を目的として開発された hFasLECD 融合蛋白質についての主な例を表1にまとめて示す[37-50]。
2. hFasLECD 誘導体の Pichia pastoris 分泌発現生産系を用いた調製
2.1 分泌発現生産系の構築
筆者が本研究を開始する以前に、P. pastoris を用いた hFasLECD の分泌発現生産に関する先行研究として、大阪バイオサイエンス研究所と持田製薬株式会社の共同研究グループから、完全長の当該ドメインに対応する103番目から281番目までのアミノ酸残基を対象とした P. pastoris GS115 株を宿主とする研究結果が発表されていた[19]。1997年に J. Immunology 誌に発表されたこの研究で用いられた hFasLECD の分泌発現用遺伝子ユニットは、5’ 末端側から P. pastoris 由来のアルコールオキシダーゼ1(AOX-1)遺伝子のプロモーター配列、パン酵母(Saccharomyces cerevisiae)由来のα-接合因子遺伝子の分泌用プレプロシグナル配列、ヒト由来の cDNA を鋳型とし PCR 法により複製増幅された DNA フラグメントにより構成されている。またこの遺伝子ユニットを搭載するベクターとしてはヒスチジンに関する栄養要求性型選択マーカー遺伝子(HIS4)を含むプラスミドの一つである pPIC9 が用いられた。発現誘導の条件としては、P. pastoris を宿主としメタノールにより発現の誘導を行なう場合の代表的な培地である Buffered Methanol-complex Medium(通称:BMMY 培地)が使用され、30℃、48時間の培養条件下で OD600 = 100 という高濃度の培養開始時菌体密度条件下に実施された。この際の分泌生産量については、後に P. pastoris による蛋白質の発現生産に関する権威の一人である J.M. Cregg 博士らにより2000年の FEMS Microbiology Reviews 誌に発表された総説[51]の中で、100 mgL-1 という培養液の単位容積当りの生産量としてかなり高いレベルの値が記載されているが、これは先に述べた菌体密度の高さが一因となっているのかも知れない。以下に述べる研究はこの先行研究の結果を基盤として行ったものである。
筆者らの研究では、化学合成遺伝子を使用してヒト由来の溶菌酵素の一種であるヒトリゾチーム[52]ならびに小麦由来のレクチンの一種である小麦胚芽アグルチニン[53]を対象とした酵母を宿主とする異種蛋白質の分泌発現生産に関わってきた経緯から、hFasLECD の P. pastoris を用いた発現生産系の構築における構造遺伝子の調製においても同じく化学合成法を採用した[20]。この構造遺伝子の設計に当たっては酵母において多量に発現される同種蛋白質遺伝子において優先的に使用されているコドン[54]を主に使用した。一方、上記の先行研究の場合と同じく、宿主としては P. pastoris GS115 株を使用し、AOX-1 プロモーター配列とα-接合因子遺伝子のプレプロシグナル配列の後に野生型 hFasLECD の103番目から281番目までのアミノ酸残基をコードする遺伝子を連結させたユニットを構築した。構築した発現用遺伝子ユニットを抗生物質ゼオシン(Zeocin)に対する抵抗性遺伝子を選択用マーカーとして含む分泌発現用ベクターの一種である pPICZαA に搭載したものを用いて発現試験を行った[20]。発現の誘導は同じくBMMY 培地を使用し、29.5℃で実施した。残念ながら発現誘導開始時の菌体密度に関する詳細な記録が残っていないため正確に比較することは困難であるが、培養上清中の分泌生産レベルは 1〜2 mgL-1 程度と先行研究の場合と比べてはるかに低いものであった。
2.2 タグ配列付加ならびに糖鎖付加部位欠失の影響
発現生産量を上昇させるための最初の試みとしてタグ配列の付加を行った。発現産物の精製工程においてアフィニティークロマトグラフィーを利用する可能性を考慮して、FLAG(AspTyrLysAspAspAspAspLys)タグもしくは (His)6 タグを選択し、かつそれぞれに精製時の立体障害に起因する収量低下を回避するべく (Gly)5 からなるフレキシブルなスペーサー配列を介して構造遺伝子の N 末端側あるいは C 末端側に接続した計4種類のタグ付加型誘導体遺伝子を作成して分泌発現を試みた[20]。その結果、タグ配列を付加しない場合と比較して、N 末端側に FLAG-(Gly)5 配列を配置した場合にのみ分泌生産量の向上が認められた(図4)。この事例を含めて、以降本研究で検討した hFasLECD の各種誘導体における構造遺伝子ユニットの構成と培養上清の SDS-PAGE 分析により推定されたおよその分泌生産レベルの増減の関係を図4にまとめて示した。次に hFasLECD 遺伝子中に3箇所存在する N 結合型糖鎖付加部位を構成する Asn 残基(Asn184、Asn250 及び Asn260)に関する改変を試みた。 N 末端に FLAG-(Gly)5 タグ配列を付加した誘導体について N 末端側から順に1箇所、2箇所ならびに3箇所の Asn 残基をそれぞれ Gln 残基に変換して糖鎖付加部位を欠失させたものを作製し、野生型とともに SDS-PAGE 分析で目的とする発現産物のバンドの濃さにより比較したところ、野生型を含めた分泌生産量の比較では Asn184Gln 変異体が最も多く、精製試料の収量は約 5 mgL-1 となることが明らかとなった。
一方、同じくN末端側に付加した場合でも (His)6-(Gly)5 タグでは逆に分泌生産量は半減した。また、いずれのタグ配列でも C 末端側に付加した場合や3箇所の糖鎖結合部位の全ての Asn 残基に Gln 残基への変異を導入した場合には、目的とする発現産物の分泌生産は認められなくなり、これらの場合には分泌過程での hFasLECD を構成するペプチド鎖の正常な立体構造形成への重大な悪影響が生じていることが推測された。特に Asn260Gln 変異の導入効果は大きく、同じく1箇所の糖鎖結合部位でも Asn260 残基のみを残存させた場合には変異導入前の場合とほぼ同程度の分泌生産量が認められたのに対して、Asn250 残基のみを残存させた場合には5個の独立した形質転換体のシングルコロニーの培養上清を SDS-PAGE を用いて解析した限り、いずれについても目的物の生産量は極めて少ないか、あるいは一旦対応する発現産物の薄いバンドが出現したように思われるものでも培養を続ける間に消失し、分解や不溶化を起こしやすい少量の発現産物が分泌されたものと推測された(図4、未発表データ)。これらの結果は P. pastoris の分泌経路内での hFasLECD の正常な立体構造の形成や発現産物の安定性の維持に関して Asn260 への糖鎖の付加が極めて重要であることを現している。また、各部位に糖鎖が付加したものに対応する発現産物の SDS-PAGE 分析におけるそれぞれのバンドの濃さから判断して、少なくとも P. pastoris GS115 株を宿主とした場合には、糖鎖付加に関して Asn184 部位の欠失の影響は小さく、この部位に糖鎖が付加する割合は少ないこと、一方 Asn250 部位の欠失の影響は大きく、この部位への糖鎖付加はほぼ完全に生じることが明らかになった[20]。以上から、同じN結合型糖鎖付加部位でも立体構造上の位置の違いに応じて P. pastoris を宿主とした分泌発現生産に与える特徴や挙動が大きく異なることが示唆された。
hFasLECD の P. pastoris を宿主とした分泌生産において Asn260 に付加される糖鎖の構造的完全性が重要であることを探るために、BioGrammatics 社から市販され SuperMan5 株と名付けられている、酵母でのコア糖鎖部分から外側への糖鎖の伸長に関わる OCH1 遺伝子が欠損し Man5 と呼ばれるコア型糖鎖(Man5GlcNAc2)部分のみが付加される株[55]を宿主とする分泌発現を試みた。この SuperMan5 株を用いた系では、2.3項で後述する GS115 株を用いた系で最も分泌発現生産量が多かった変異体の一つである [Δ(103-138),Asn184Gln+Asn250Gln] 変異体について検討した場合でも、培養液を直接 SDS-PAGE で分析した限り明確な発現産物のバンドを検出することはできなかった(図4)(未発表データ)。この結果は、Asn260 位に付加するコア構造以外の部分を含めた糖鎖構造の完全性が P. pastoris の分泌経路における hFasLECD 分子の正常な立体構造形成の過程において極めて重要であり、その過程においてグルコシダーゼ I および II ならびにカルネキシン、カルレティキュリンなどのシャペロン機能を有するレクチン様分子が関与するコア型構造部分以外の糖鎖に対する認識[56]が本質的な役割を果たしていることを強く示唆している。不完全な構造の糖鎖を有するために正常な立体構造が形成されなかった hFasLECD 分子は、おそらく小胞体(ER)内での品質管理機構である小胞体関連分解(ER-associated degradation)による処理を受けリサイクル過程に回されたものと考えられる[57]。
当初、著者は hFasLECD 単体での立体構造解析のための結晶化を目指していたため、Asn184 残基のみ、あるいは Asn184 残基と Asn250 残基を同時に Gln に変換し N 結合型糖鎖付加部位をあらかじめ部分的に欠失させた変異体について、さらに化学構造の均一化を図るべく、Endo Hf グリコシダーゼ(NEB 社製)による残存する糖鎖のトリミングを行い、単一の N-アセチルグルコサミン残基への変換を試みた[58]。結果として、Asn250 残基に結合している糖鎖は短時間で容易に切断されるのに対し、Asn260 残基に結合した糖鎖の切断にはより多くの酵素量と反応時間を要することが明らかになった。この事実は先に述べた Asn260 残基に付加する糖鎖の hFasLECD の立体構造安定性への正の寄与と呼応するものである。また、[Δ(103-138),Asn184Gln+Asn250Gln] 変異体に残存する Asn260 残基に付加された糖鎖を Endo Hf で切断し N-アセチルグルコサミン1残基のみとしたものについて、結晶化条件探索のための実験に必要な濃度までの濃縮を試みたところ、一定の濃度に達した段階で沈殿物が生じ、不溶化を伴う凝集が起こりやすい生成物であった(未発表データ)。この現象は大腸菌菌体内で生産された糖鎖を持たない hFasLECD の発現産物を再構成して可溶化した試料が凝集を起こし易いという観察[10]とも一致している。残存する糖鎖を Endo Hf で切断した精製試料について MALDI-TOF 法による質量分析を用いた解析(アプロサイエンス社受託)を行ったところ、確かに予想された N-アセチルグルコサミン1残基のみが付加された均一な化学構造と一致した分析結果が得られたことから、P. pastoris GS115 株を宿主として分泌生産された発現産物は O 結合型糖鎖の付加などの他の翻訳後修飾は受けていないものと推察された[58]。
2.3 N 末端領域の除去が分泌生産量に与える効果
hFasLECD の構造遺伝子については、過去に哺乳動物細胞を用いて行われていた一連の欠失変異体の解析[59]から、N 末端部位のアミノ酸残基103番から138番目までの領域は FasR 結合活性の発現に必要な3量体構造を形成する上においては必須ではないことが既に明らかになっていた。この領域を除去して P. pastoris を宿主とした分泌生産を行った場合にどのような影響が生じるかを明らかにする目的で、上述した Asn184Gln 変異体ならびに [Asn184Gln+Asn250Gln] 二重変異体をベースとしてさらにこの領域に欠失変異 [Δ(103-138)] を追加した遺伝子を作成して発現生産を試みた[58]。その結果、興味深いことにいずれの変異体の場合にも大幅に分泌生産量が増加し(図4)、[Δ(103-138),Asn184Gln+Asn250Gln]変異体の場合、その精製収量はおよそ 24 mgL-1 に達することが明らかとなった。当該欠失領域の C 末端にはPro134Ser135Pro136Pro137Pro138 というプロリン残基が多数連続するアミノ酸配列が存在(図2)しており、その特異性から正常な立体構造を形成する際の律速段階となっている可能性がある。[Δ(103-138)] 変異体ではこの配列が除去されたために、P. pastoris の ER 内での効率的な立体構造形成が起こり大幅な分泌生産量の増加につながったのではないかと考えられる。
2.4 陽イオン交換クロマトグラフィーによる精製
2.1項で述べた P. pastoris を宿主とした先行研究においては、分泌生産された発現産物は高マンノース型糖鎖が付加された糖蛋白質であることを利用して、精製工程の第一段階に Con A アガロースによるアフィニティークロマトグラフィーが使用された[19]。一方、著者は研究開始当初から糖鎖のトリミングによる化学構造の均一化を計画していたため、原理的に糖鎖の有無に依存しない陽イオン交換クロマトグラフィーによる分泌発現産物の精製法を開発した。
野生型 hFasLECD の一次構造中には酸性アミノ酸残基である Asp 残基と Glu 残基を合わせた数に比べて塩基性アミノ酸残基である Lys 残基と Arg 残基を合わせた数の方が多く含まれ、アミノ酸残基103番から281番までの領域には、酸性残基が16個と塩基性残基が20個、アミノ酸残基139番から281番までの領域には、酸性残基が12個と塩基性残基が17個存在する(図2)。ExPASy Proteomics Server[60]を用いた計算によると等電点(pI)はそれぞれ理論上8.96ならびに9.15と予測された。予備実験の結果、幸いにして調製した各変異体は pH 5.5 付近でも安定に可溶性で存在することが明らかになったため、スルホプロピル基が坦持された陽イオン交換体である Hi-Trap S カラム、Resource S カラムあるいは Mono S カラム(いずれも GE ヘルスケア社製)を用いた分画精製を行った。具体的には培養上清を濃縮後、pH 5.3~5.6 の 50 mM 酢酸ナトリウム緩衝液への充分な置換を行った試料について、最初に Hi-Trap S カラムを用いたマニュアルのシリンジ操作での塩濃度を段階的に増加させた溶出による粗分画を行い、引き続きより分離能が高い Resource S カラムまたは Mono S カラムを用いた高速液体クロマトグラフィーシステムによる塩濃度を連続的に上昇させた溶出による分画を実施して、最終精製試料を調製した。
N 末端領域を欠失させ、かつ N 結合型糖鎖が付加される残基を Asn260 のみとした [Δ(103-138), Asn184Gln+Asn250Gln] 変異体は、精製工程におけるクロマトグラム上3箇所の N 結合型糖鎖付加部位を有する野生型の試料に比べてよりシャープな形状の目的物のピークが得られたことから、分泌生産量が多くより純度の高い試料の調製に有利であることとも併せて、以後の実験においてはこの変異体を各種誘導体作成のための基本的な変異体として使用することにした。また、陽イオン交換クロマトグラフィーを用いる精製に関連して、3.1項以降で詳述する部位特異的化学修飾体の調製を行った際に、一部のものについては、N 末端に付加した FLAG-GlyCys(Gly)4 タグ配列中に塩基性残基である Lys を新たに3残基挿入した FLAG-(Lys)3GlyCys(Gly)4 タグ配列を使用することにより、上記の Hi-Trap S カラムを用いた粗精製段階における分画前試料に関する緩衝液の置換をより簡略的に行えるようにすることで、精製工程における操作性を向上させることが可能であった[61]。
2.5 ヒト Fas レセプター細胞外ドメインに対する結合活性の確認
hFasL による標的細胞に対するアポトーシスの実行は、この蛋白質分子に対して現在知られている唯一のシグナル伝達実行型の受容体であるヒトFasレセプター細胞外ドメイン(hFasRECD)との特異的な結合を介して誘起される。したがって、P. pastoris を宿主として分泌生産された hFasLECD の誘導体ががん細胞などの標的細胞に対してアポトーシスによる細胞死誘導活性を示す潜在的能力を有するかどうかについては、基本的に hFasRECD に対する特異的な結合活性の有無により判断することが可能であり、本研究においてもこの結合活性評価試験用に適した hFasRECD を含む分子が必要であった。しかしながら、その目的に合う代表的な分子である hFasRECD とヒト IgG1 の Fc ドメインの融合蛋白質(hFasRECD-Fc)に関しては、研究用試薬としてマウス由来の培養細胞の一つである NS0 細胞を用いて発現生産された精製試料(R&D 社製)などが当時市販されていたものの、いずれも少量かつ高価であったため必ずしも容易に使用できる状況にはなかった。そのため、自ら試験用の試料を調製する必要に迫られ、先に2.1項で述べた hFasLECD 遺伝子を同じく酵母における頻用コドンを用いて化学合成により調製した hFasRECD 遺伝子と置き換えた形の発現用遺伝子ユニットを構築し、同じ手順により分泌発現を試みたが期待に反して培養液中に目的の発現産物の分泌を確認することはできなかった(未発表データ)。
そこで、先行研究として文献上の記載があったマウス由来の FasRECD とヒト IgG1 の Fc ドメインの融合蛋白質の例[62]にならい、昆虫細胞を使用した発現生産系[63]を用いて hFasRECD-Fc を調製する方策に転換した。その際には、有償の受託発現サービスを利用した。最初に、まず発現生産用に用いられる代表的な昆虫培養細胞の一つであるヨトウ蛾(Spodoptera frugipera)由来の sf9 細胞を用いる系[64](タカラバイオ社受託)で比較的高収量(6.7 mgL-1)の分泌発現が可能であることを確認した後、より生産量の増加が期待できるカイコ蛾(Bombyx mori)の幼虫の血リンパ液中への分泌生産[65](片倉工業社受託)を試みた。後者の系は良好に機能し、発現生産のためのバキュロウイルスを感染させた5齢幼虫由来の 26 ml の血リンパ液から 22.5 mg の精製試料が得られた。尚、カイコ蛾幼虫の血リンパ液中には内在性の夾雑蛋白質が多量に存在するため目的とする組換え体蛋白質の精製が困難になる場合があることが予想されるが、この点に関しても hFasRECD-Fc の場合、当該 Fc ドメインに対する高い特異性と充分な結合容量を兼ね備えたプロテイン G を坦持した市販のアフィニティー精製用カラム(Hi-Trap プロテイン G、GE ヘルスケア社製)が入手可能であったため、これに引き続く高分離能の陰イオン交換クロマトグラフィー用カラム(Resource Q、GE ヘルスケア社製)を用いた分画と組み合わせることにより簡便に精製試料の調製を行うことができた[66]。
hFasLECD の hFasRECD に対する特異的結合活性の評価試験での利用において hFasRECD-Fc の有利な点の一つは、市販のプロテイン A またはプロテイン G が坦持されたアガロースビーズや磁気ビーズを併せて用いることにより、共免疫沈降試験が実施可能であることである。この試験において、これまでに調製した hFasLECD の誘導体は後述する部位特異的化学修飾を施したものも含めていずれも hFasRECD-Fc と安定な複合体を形成して沈降物中に回収され、hFasRECD に対する強い結合活性を有することが確認されている。
また、hFasLECD と Fc ドメインが結合していない hFasRECD との複合体の形成について調べるため、hFasRECD-Fc における両ドメイン間のヒンジ領域にセリンプロテアーゼの一種であるトロンビン(Thrombin)による特異的認識切断サイトとして AlaAlaAlaProArgGlySerAla の配列を挿入した hFasRECD-T-Fc 型誘導体についてもカイコ幼虫を用いた分泌発現生産を行った[67]。この hFasRECD-T-Fc についても高レベルの発現生産が認められ、組換えカイコ幼虫由来の 25 ml の血リンパ液から 13.5 mg の精製試料が得られた。これをトロンビンにより切断後、切り離されたFcドメインをプロテイン G 坦持カラムにより吸着除去し、さらに素通り画分中に含まれる蛋白質を陽イオン交換カラム(Resource S)により分画することにより、hFasRECD 部分のみからなる精製試料を調製した。尚、hFasRECD 中にはトリプトファン残基が含まれないため、P. pastoris を用いて生産された hFasLECD 誘導体の単離された hFasRECD に対する溶液中での特異的結合については、両者を種々の割合で混合した試料についてのサイズ排除クロマトグラフィーを用いた分析を行い、215 nm と 280 nm の2種類の波長の吸光度を同時に検出することで確認した[67]。
さらに、hFasRECD-T-Fc 分子内の hFasRECD 中に存在する2箇所の N 結合型糖鎖付加部位に関してそれらを構成する Asn 残基を共に Gln 残基に変換することで欠失させた変異体についても、発現量の低下は認められたものの、同様にカイコ幼虫の血リンパ液中への分泌生産により調製することが可能であった[68]。また、N 末端タグ配列が付加されていない hFasLECD の [Asn184Gln+Asn250Gln] 二重変異体中に残存する糖鎖を Endo Hf を用いてトリミングした試料と上記の hFasRECD ドメイン領域内に糖鎖付加部位を持たない hFasRECD-Fc 変異体試料を用いて共免疫沈降試験を行った場合にも、当該リガンド–レセプター間の強い結合が確認された。この結果は、少なくとも試験管内での hFasLECD と hFasRECD の結合においてはそれぞれが有する糖鎖に由来する相互作用の寄与は少ないことを示唆している。
2.6 ディスポーザブルカルチャーバッグを用いた培養システムの開発
P. pastoris を宿主とする異種蛋白質の分泌生産実験においては、発現誘導用として P. pastoris 由来の AOX-1 遺伝子の強力なプロモーター遺伝子領域を、また分泌用シグナルとして S. cerevisiae 由来のα-接合因子遺伝子のプレプロシグナル配列を使用することが効率的な分泌発現生産用遺伝子ユニットを構成する上での代表的な戦略の一つになっている[11]。P. pastoris はメタノール資化性の酵母であり、メタノールを唯一の炭素源とした培養が可能である。その場合、代謝の第一段階であるメタノールのホルムアルデヒドへの変換のために多量のアルコールオキシダーゼが必要となるため、自ずから AOX-1 プロモーターの転写活性が高まる。また、その変換反応における基質の一つである酸素分子は、培地に対する溶解度が低くアルコールオキシダーゼに対する親和性は必ずしも高くない[69]ため、培養時の通気を良くすることによりその充分な供給を確保することが遺伝子発現の効率を高める上で極めて重要になる。
hFasLECD の発現生産系を構築するに当たって P. pastoris を宿主とする分泌発現系を選択した理由の一つに、小規模な研究室において使用可能なレベルの装置や培養規模でも立体構造解析のための結晶化探索実験や部位特異的な化学修飾を手段とする高機能化を目指した実験に必要な量の精製試料を調製できる可能性があることが挙げられる。本研究では、市販の微生物培養用の全容積 5 L あるいは 10 L のポリプロピレン製のディスポーザブルカルチャーバッグ(藤森工業社製)を培養容器として用い、ダイアフラム式の小型ポンプにより強制的な通気を実施する培養システムを新たに開発して使用した(図5a、5b)[68]。このシステムにおいて、実際に使用可能な培地量は各カルチャーバッグ全容積の半分以下であるため、上記のバッグを使用した場合、それぞれ一回で 2.5 L あるいは 5 L 規模までの培養が実施可能である。それまでに使用してきた 3 L 容量のガラス製バッフル付きフラスコ(培養液量、500 mL/本)を用いた培養システムとカルチャーバッグを使用するシステムでの分泌生産量を、N 末端にタグ配列が付加されていない hFasLECD の [Δ(103-138),Asn184Gln+Asn250Gln] 変異体を対象として最終的な精製物の収量で比較したところ、単位培養容積当り約3倍の増加が認められた[68]。この培養システムは、これまでフラスコ培養の際に使用してきた恒温回転振とう式の培養機をそのまま利用可能で、発現誘導用の培地についても同じ組成のものが使用できるためガラス製バッフル付きフラスコを用いた系から簡便に移行することができる。また、本研究で使用したディスポーザブルカルチャーバッグは比較的安価(5 L 容量および 10 L 容量共に2万円弱/5セット)で入手可能であった[17]。次項で述べる部位特異的化学修飾体の調製実験では反応に供するためにできるだけ多量の N 末端タグ配列中にシステイン残基を導入した誘導体の精製試料が必要であったため、その取得を目的とした発現生産実験においては専らこのシステムを使用した。陽イオン交換クロマトグラフィーを用いた精製工程に供するための培養上清中の目的とする発現産物の効率的な濃縮ならびに脱塩操作においては、並列に連結した複数個のペリスタ式ポンプのそれぞれにポリエーテルスルホン製の限外濾過膜(排除限界分子量: 10 kDa)を内蔵したタンジェンシャルフロー(TFF)型の小型カセット(ポール社製)を接続した装置の利用が有効であった(図5c)。
3. hFasLECD の部位特異的化学修飾体の調製とキャラクタリゼーション
3.1 N 末端タグ配列中にシステイン残基を有する誘導体の設計と発現生産
hFasLECD の部位特異的化学修飾を手段とする高機能化の戦略を考えるに当たっては、化学修飾の導入後もこの蛋白質の医療応用上最も有用な生物活性の一つである hFasRECD に対する特異的な結合に基づくアポトーシス誘導活性を保持させることを念頭に置く必要がある。そのためには、hFasRECD との結合の際の相互作用様式を事前に立体構造の観点から検討しておくことが重要である。1.1項で述べたように、一次構造の相同性から hFasLECD-hFasRECD 複合体と立体構造が類似していると推定されるデスリガンド‐デスレセプターの組み合わせであるヒト TNFβ-ヒト55kd TNF レセプター細胞外ドメイン複合体の X 線結晶構造解析結果[70]などをもとに、hFasLECD(サブユニット3量体)は hFasRECD(単量体)と1:3のモル比で複合体を形成することが比較的古くから推定されていた。この推定の正しさは、hFasLECD とデコイ型レセプターである hDcR3 の複合体の立体構造が発表されるに至り[10]、ほぼ確定的になったものと考えられる。
この複合体の立体構造解析結果によると hFasLECD の N 末端部位は hDcR3 との結合の際に互いが直接接触する界面部位から三次元的に離れた位置に存在している(図3)。そのため、N 末端に親水性の高いタグ配列を付加した場合、タグ配列を構成するポリペプチド鎖は hFasRECD に対する結合を阻害することなく溶媒中に露出して存在することがあらかじめ予測できた。この予測に基づいて、2.2項で記した hFasLECD 本体(アミノ酸残基番号139-281)の N 末端に付加した FLAG-(Gly)5 タグ中の連続した Gly 残基の領域内に部位特異的化学修飾反応の足がかりとなるシステイン(Cys)残基を挿入することにより FLAG-GlyCys(Gly)4 とした。その際に Cys 残基を最初の Gly 残基のあとに配置した理由は、当該 Cys 残基に対して化学修飾により付加される分子が FLAG タグ配列固有の機能になるべく影響を及ぼすことなく、かつ挿入した Cys 残基が hFasLECD 本体からできるだけ離れた位置にくるようにすることで溶媒中に露出しやすくなり化学修飾に関する反応性が高まることを期待したためである。
この N 末端タグ配列内に Cys 残基を導入した誘導体について、当該 Cys 残基を含まない誘導体の場合と同一の方法を用いて P. pastoris GS115 株を宿主とした発現を試みたところ、3量体中の2つのサブユニットの一部がそれぞれのタグ配列内に存在する Cys 残基の間でジスルフィド(SS)結合を形成して培養液中に分泌されることが非還元条件下での SDS-PAGE 分析により明らかになった[68]。しかし、この部分的にサブユニット間の SS 結合が形成された発現産物についてもそれまでのタグ配列内に Cys 残基を含まない誘導体の場合と同様な方法で精製をすることが可能であった。
野生型の hFasLECD 分子中には立体構造上 Cys202-Cys233 間にサブユニット当り1か所の SS 結合が元来存在する(図3)。そのため、このドメイン内部に埋もれた SS 結合を保持した状態でN末端部位に連結したタグ配列内に存在する Cys 残基に対して部位特異的にかつ効率的に化学修飾を施すためには、タグ配列中の Cys 残基の側鎖のみを反応性の SH 基を持つように活性化した状態に保つ必要がある。この目的を達成するためにはトリス(2-カルボキシエチル)ホスフィン(TCEP)の使用が有効であった。TCEP はその比較的大きな分子サイズのため、2-メルカプトエタノール(2ME)やジチオスレイトール(DTT)などのチオール性還元剤を使用した場合に比べて、適切な濃度や反応温度を設定することで、蛋白質分子内部に埋もれた SS 結合に影響を与えることなく、より選択的に溶媒中に露出した SS 結合のみを還元して反応性を有する SH 基とすることができる[71]。また、反応機構上も 2ME や DTT を用いた場合のような SS 結合の交換反応が起こらないため、基本的には過剰の TCEP を除去することなくマレイミド化合物等による修飾反応に供することが可能である。P. pastoris を宿主として分泌生産された上記の N 末端タグ配列内に Cys 残基を含む誘導体の場合、室温(25℃)、pH 5.5 の条件下で、10~20 mM の TCEP を1時間程度作用させることで選択的な SH 基の活性化の目的を達成することができた。他方、過剰の TCEP がマレイミド基を持つ化学修飾用試薬と反応し、ペプチド中に含まれる Cys 残基とマレイミド基を有する色素分子との反応を阻害する可能性を指摘する報告もある[72]。そのため、次の段階の修飾反応に移る前に、マレイミド化合物との反応に適した pH 6.4 の 25 mM のリン酸緩衝液への交換を兼ねて、念のため事前に PD-10 カラム(GE ヘルスケア社製)を用いた分画により余分な TCEP をできるだけ除いた直後にマレイミド化合物との反応に供した。その際に 2~5 mM 程度の EDTA を共存させることにより SS 結合の再生を防いだ。尚、過剰の TCEP を除いた後の試料を非還元条件の SDS-PAGE 分析に供した際に、SS 結合が再生された産物に対応すると考えられる分子量の位置に薄いバンドが認められたが、上記のマレイミド化合物との反応後にはこのバンドは確認できなかった[68]ことから、このバンドは SDS-PAGE 分析の操作過程において pH の上昇などが原因で SS 結合が一部再生することにより検出されたものと推察された。
3.2 部位特異的化学修飾体の調製法の検討
これまでに hFasLECD に関する部位特異的化学修飾体(部位特異的化学修飾による他分子との共有結合体)の調製法として、上記のタグ配列中に Cys 残基が含まれる誘導体を対象として、i) 大過剰の目的とする修飾用分子のマレイミド基含有誘導体をチオール–エン反応により直接付加させる方法[73]、ならびに ii) 最初に6-メチルテトラジン(MTZ)基またはトランスシクロオクテン(TCO)基を含むマレイミド化合物とのチオール–エン付加反応により末端に TCO 基あるいは MTZ 基を持つスペーサー用分子を結合させた hFasLECD 誘導体を調製し、一旦単離した後、引き続き目的とする修飾用分子との間で TCO 基–MTZ 基間の逆電子要請ディールスアルダー型(iEDDA)付加反応により連結を行う方法[61]の2種類について検討した(図6)。図7に本研究において各蛋白質分子に対して末端に TCO 基ならびに MTZ 基を含むエチレングリコールオリゴマー型スペーサーを導入する際ならびに付加反応後残存する未反応の TCO 基および MTZ 基のクエンチング反応を行う際に使用した市販試薬(いずれも Click Chemistry Tools 社製)の化学構造式を示した。また、引用文献61の Fig.1 にこれらの試薬を用いて調製した TCO 基あるいは MTZ 基を末端に有する各タンパク質分子に関する誘導体の構造式が記載されているので併せてご参照いただきたい。
化学修飾法による分子の付加は必ずしも定量的に達成できるとは限らない。チオール–エン反応による直接付加の事例として、N 末端に FLAG-GlyCys(Gly)4 タグ配列を有する hFasLECD の [Δ(103-138),Asn184Gln+Asn250Gln] 変異体に対して大過剰のフルオレセイン-5-マレイミドを用いて反応を行なった際の修飾率は精製試料中に導入された蛍光色素団固有の吸光度値から83%と推算された[73]。一方、iEDDA 付加反応を用いた事例として、N 末端に FLAG-(Lys)3GlyCys(Gly)4 のタグ配列を持つ hFasLECD の [Δ(103-138),Asn184Gln+Asn250Gln] 変異体に対して、20倍過剰モル当量の3単位のエチレングリコールユニットの両端に TCO 基とマレイミド基を有する化合物(TCO-PEG3-MAL)(図7)を反応させて調製した hFasLECD の TCO 基含有誘導体(hFasLECD-PEG3-TCO)と MTZ 基ならびにメトキシ基を末端に持つ分子量約 5 kDa のポリエチレングリコール(PEG)の誘導体(mPEG-MTZ, 5 kDa)(図7)との反応を実施した。この付加反応においては1.0~1.5倍過剰モル当量程度の mPEG-MTZ, 5 kDaの使用量で付加体の収率は飽和に達してほぼ一定になることが反応混合物の SDS-PAGE 分析により確認されたが、この場合にも修飾率は80%程度であった[61]。
通常マレイミド基を含む修飾用分子を直接用いる反応の場合は被修飾蛋白質分子に対して過剰なモル比の修飾用試薬が必要になるが、1段階反応での付加が行なえるため全体として目的とする分子による修飾率が2段階反応を行う場合に比べてより高くなる。そのため、修飾用分子のマレイミド誘導体が比較的容易に入手可能な場合に適している。一方、第2段階として使用される TCO 基–MTZ 基間の付加反応は Cys 残基–マレイミド基間の反応を用いる場合に比べて、被修飾分子に対してより少ないモル比の修飾用分子を用いても室温下で円滑に反応が進行する。そのため、より貴重な低分子化合物や機能性蛋白質分子など一般的には潤沢な量が得難い分子を被修飾用分子としたい場合に適していると考えられる。具体的には、高い修飾率を目指して反応を行ないたい場合、Cys 残基とマレイミド基の反応においては典型的には25倍過剰モル当量程度の修飾用試薬を使用する必要がある[71]のに対し、TCO 基と MTZ 基の付加反応の場合には 1~数 mgmL-1 程度の濃度の蛋白質分子の溶液について1.0~1.5倍過剰モル当量程度の被修飾分子を添加した場合でも高い修飾率の達成が期待できる。
また、反応前の試料を保存する際に、Cys 残基やマレイミド基の場合は通常多くの蛋白質分子が安定に存在できる pH 7~8 付近の生理的緩衝液条件下において、それぞれ空気酸化による SS 結合の形成ならびに加水分解によるマレアミド酸基への変換が起こりやすく、これらに由来する官能基の不活性化の可能性を常に考慮する必要がある。これに対し、TCO 基や MTZ 基の場合には同様な条件下での安定性が高く、通常は試料溶液を-20℃以下で凍結保存すれば長期間にわたって大きな活性の低下を危惧せずに使用可能である。そのため、モノクローナル抗体などを始めとする貴重な蛋白質を修飾用分子として用いる場合には特に有用であると考えられる。
3.3 低分子性蛍光色素による化学修飾体の調製
これまでに上述した N 末端タグ配列中に Cys 残基を導入した2種類の hFasLECD 誘導体に関する低分子性蛍光色素による化学修飾体として、フルオレセイン-5-マレイミドを用いた1段階反応法による修飾体の調製[73]、ならびに TCO 基–MTZ 基間の付加反応を用いた2段階反応法によるスルホ-Cy3 基を導入した修飾体の調製[61]を行った。調製した全ての蛍光性化学修飾体は hFasRECD-Fc(または hFasRECD-T-Fc)とプロテイン A 担持アガロース(ロシュ社製)(またはプロテイン G 坦持磁気ビーズ、バイオラッド社製)を使用した共免疫沈降試験により、非修飾体と同等な hFasRECD 結合活性を有することが明らかとなった。加えて、各蛍光色素団に由来する可視領域の吸収極大(フルオレセイン基:495 nm、スルホ-Cy3 基:550 nm)付近の吸光度を指標とするサイズ排除クロマトグラフィーを用いた複合体形成確認試験においても明確な hFasRECD 結合活性が確認できた。これらの結果は、先述した hFasLECD–hDcR3 間複合体の立体構造解析情報に基づく部位特異的化学修飾の戦略が hFasRECD 結合活性の保持において有効であったことを示している。
調製した蛍光性化学修飾体はいずれもそれぞれの蛍光色素団に特徴的な励起ならびに蛍光スペクトルを示したが、精製物試料の紫外・可視領域の吸収スペクトルから推算された hFasLECD サブユニット3量体当りの各蛍光色素基の平均導入数は、1段階反応により導入したフルオレセイン基の場合2.5であったのに対し、2段階反応で導入したスルホ-Cy3基の場合には、スルホ-Cy3 基を含む分子側の官能基として MTZ 基ならびに TCO 基を用いて導入した各修飾体でそれぞれ1.5および1.6であり、あらかじめ予想されたように付加反応の段階数の増加による蛍光色素基の導入率の低下が認められた。また、サイズ排除クロマトグラフィーを用いた各蛍光性化学修飾体と hFasRECD-Fc との複合体形成試験において、TCO 基含有誘導体の形でスルホ-Cy3 基を導入した修飾体では、同様な分析条件にもかかわらず、上記のフルオレセイン基修飾体や MTZ 基含有誘導体の形でスルホ-Cy3 基を導入した蛍光性修飾体を用いた場合に比べて、hFasLECD の蛍光色素修飾体と hFasRECD-Fc の複合体がカラム内でわずかながら解離しやすい傾向が認められた[61,73]。この結果は TCO 基含有誘導体の形でスルホ-Cy3 基を導入した場合には、修飾により hFasLECD と hFasRECD-Fc の相互作用における会合強度が立体障害等による何らかの理由により低下した可能性を示唆するものである。そのため、次項で述べる他の機能性蛋白質分子を修飾用分子として用いた架橋体の作成実験においては、修飾用蛋白質分子の MTZ 基含有誘導体と hFasLECD-PEG3-TCO の間の iEDDA 付加反応を使用した。
3.4 他の機能性蛋白質分子との架橋体の調製
筆者はこれまでに hFasLECD-PEG3-TCO(約 60 kDa)に関する他の機能性蛋白質による修飾のモデル実験として、ニワトリ卵白由来のアビジン(Avi、約 66 kDa)ならびにウサギ IgG 由来の Fab’ ドメイン(rFab’、約 55 kDa)を使用した架橋体の調製について検討した[61]。図8aに hFasLECD とこれらの蛋白質のおよその分子の大きさと形の比較のために PDB に登録されているデータをもとに描画した立体構造(但し、rFab’ については Fab 部分のみ)を示した。
4つの等価なサブユニットからなる Avi 分子中には1サブユニットあたり Lys 残基が9個存在する。Avi に関する MTZ 基含有誘導体(Avi-PEG4-MTZ)は、サブユニット当り8倍過剰モル当量の4単位のエチレングリコールオリゴマーユニットの両端に MTZ 基ならびにスルホ-N-ヒドロキシスクシンイミドエステル基を有する化合物(MTZ-PEG4-sulfo NHS ester)(図7)を 0.1 M の炭酸水素ナトリウム水溶液(pH 8.3)中、室温(24℃)で反応させることにより調製した。そのため、原則的には分子表面に存在する不特定な Lys 残基の位置に MTZ 基が導入されると考えられる。尚、Lumiprobe 社の技術資料 “NHS ester labeling of amino biomolecules”[74]によると、このような反応条件を用いた場合一般的な蛋白質やペプチドでは平均1個の Lys 残基が修飾されるが、後に hFasLECD-PEG3-TCO との反応により得られた架橋体の SDS-PAGE 分析による解析結果は1サブユニットに複数の MTZ 基が結合しているものが少量含まれていることを示唆していた[61]。一方、rFab’ に関する MTZ 基含有誘導体(rFab’-PEG4-MTZ)については、rFab’ と MTZ-PEG4-MAL との反応により調製した。最初に、ウサギの血清からプロテイン A が担持されたアフィニティーカラムを用いて精製された全 IgG 分子をブタ胃由来のペプシンにより切断して F(ab’)2 ドメインとしたものをさらに37℃で 10 mM の EDTA を含む 10 mM の2-メルカプトエチルアミンを用いて処理[75]することにより、C 末端近傍に単一の反応性 Cys 残基を有する rFab’ を得た。引き続き、この rFab’ ドメインを直ちに大過剰の4単位のエチレングリコールオリゴマーユニットの両端に MTZ 基ならびにマレイミド基を有する化合物(MTZ-PEG4-MAL)(図7)と反応させることにより rFab’-PEG4-MTZ を調製した。したがって、MTZ 基の導入位置は C 末端部位に限られ、導入数は1個と考えられる。
hFasLECD-PEG3-TCO と Avi-PEG4-MTZ または rFab’-PEG4-MTZ とのいくつかの混合モル比条件下での反応物について、150 mM の NaCl を含む 50 mM Tris-HCl(pH7.5)を溶出用緩衝液とし、Superdex Increase 200 10/300カラム(GE ヘルスケア社製)を用いて行ったサイズ排除クロマトグラフィー分析に関する代表的なプロフィールを図9に示した。hFasLECD-PEG3-TCO(2.5 mgmL-1)とそれぞれ1.0、1.2、1.5及び3.0倍過剰モル当量の Avi-PEG4-MTZ(4.3 mgmL-1)を混合し、28℃で1時間静置した反応物を分析した結果、いずれの混合比の場合にも非架橋体試料の溶出位置に比べて高分子量側の領域に明確に分離したいくつかのピークが出現した。新たなピークが出現したクロマトグラム領域に関しては、1:1架橋体と考えられる最大のピークを含め、各ピーク間の相対的な高さの比や分離パターンなどのプロフィールには上記の4種類の反応条件間で本質的に大きな差は認められなかった[61](図9a)。また、上記のクロマトグラムにおいては、hFasLECD-Avi の1:1架橋体の分子量は理論上約130 kDaであるにもかかわらず分子量サイズマーカーの一つとして使用した球状蛋白質であるアルドラーゼ(158 kDa)のピーク溶出位置よりも若干高分子量側の位置にピークが認められた。これは、1:1架橋体の形状が単純な球状ではなく類似した分子量の2つの球状蛋白質分子がエチレングリコールオリゴマー鎖を含むスペーサーにより連結された亜鈴様の形状(図8b)であることに由来するものと推量される。架橋反応後 Avi 分子上に残存する未反応の MTZ 基を大過剰の TCO 基を含有する1級アミン化合物(TCO-NH2・HCl)(図7)を用いてクエンチングした試料について上記の1:1架橋体に対応するピーク画分を分取した。この分画物試料は hFasRECD-Fc に対する強い結合活性を保持すると同時に、ビオチンを坦持したヤギ由来抗ウサギIgG (H&L) 抗体(アブカム社製)を用いた共免疫沈降試験ならびにビオチン誘導体化された蛍光色素分子である ATTO495-ビオチン(ATTO-TEC 社製)やビオチン-PEG5K-フルオレセイン(Nanocs 社製)との混合物について hFasLECD-Avi に結合していない過剰な蛍光色素分子をサイズ排除クロマトグラフィー分画で除去した精製複合体試料の分光学的解析を行うことにより、強いビオチン結合活性を有することが確認できた。ビオチン誘導体化蛍光色素分子との複合体の分光学的性質に関して、前者の場合、蛍光色素団と Avi 分子の間の距離が短いため両者間の相互作用に由来すると考えられる蛍光色素団の吸収スペクトルの極大波長の変化を伴う著しい消光現象が観察された[61]。一方、後者では蛍光色素団と Avi 分子の間に約 5 kDa のポリエチレングリコール鎖によるスペーサーが存在しているためこのような現象は起こらず、元来の蛍光強度を有する複合体が形成された(未発表データ)。
他方、hFasLECD-PEG3-TCO(2.5 mgmL-1)とそれぞれ1.0、2.0、3.0及び5.0倍過剰モル当量の rFab’-PEG4-MTZ(2.3 mgmL-1)を混合し、25℃で1時間静置した反応混合物を分析した結果、Avi-PEG4-MTZ の場合と同様に非架橋体の溶出位置に比べて高分子量側の領域に新たに分離したピークが出現した。Avi-PEG4-MTZ の場合と異なる点は、rFab’-PEG4-MTZ の使用モル数を増加するにつれて最初は1:1架橋体と考えられる成分が主ピークであったものが、徐々に1:2架橋体ならびに1:3架橋体と考えられるより高分子量側のピークに置き換わっていったことである[61]。すなわち、等モル当量の rFab’-PEG4-MTZ を使用した場合には1:1架橋体のピークが主であったものが、3倍過剰モル当量を使用した場合には1:1架橋体と1:2架橋体のピークの高さがほぼ同じになり、さらに5倍過剰モル当量に増加させた場合には1:1架橋体のピークはさらに減少し1:2架橋体ならびに1:3架橋体のピークが主成分となった(図9b)。これらのうち、等モル当量ならびに5倍過剰モル当量での架橋反応を行った後、hFasLECD-PEG3-TCO 分子上に残存する未反応の TCO 基を大過剰の MTZ 基を含む一級アミン化合物(MTZ-PEG4-NH2・HCl)(図7)を用いてクエンチング反応処理を行ない、各反応混合物から、1:1架橋体単独の画分ならびに1:2架橋体と1:3架橋体が主に含まれる混合物の画分をそれぞれ分取した。hFasRECD-Fc とプロテイン G 担持磁気ビーズを用いて行なった共免疫沈降試験の結果、いずれの分画物試料についても hFasRECD に対する強い結合活性を保持していることが確認できた。また、ウサギ IgG 全分子を坦持したアガロース担体を用いてアフィニティー精製されたヤギ由来抗ウサギ IgG (H&L) ポリクローナル抗体(アブカム社製)を使用した共免疫沈降試験により、この抗体により特異的に認識されるウサギ IgG 由来の Fab’ 断片が確かに結合していることを確認した[61]。
サイズ排除クロマトグラフィーによる分析結果から、Avi-PEG4-MTZ あるいは rFab’-PEG4-MTZ のいずれを使用した場合にも hFasLECD-PEG3-TCO との室温(25~28℃)下1時間処理後の等モル反応混合物には未だかなり多量(それぞれ45%および62%)の非架橋体が残存し、後者については反応時間を2倍に延長した場合にもその割合はほとんど変化しないことを確認した。この結果はたとえ効率的なクリック化学反応を利用したとしても、高分子量の蛋白質分子間の架橋反応の場合には立体障害等の観点から反応収率には一定の上限が存在する可能性を示している。この点に関しては、両蛋白質分子間の連結に使用するスペーサー長をより長くするなどの工夫により架橋体の収量を増加させることが可能になるかも知れない。
3.5 共有結合構造の同定と細胞死誘導活性の評価
これまでに3.4項で述べた hFasLECD-PEG3-TCO(サブユニット3量体)と Avi-PEG4-MTZ(サブユニット4量体)の間の付加反応により得られた1:1架橋体(図8b)の分画物試料を用いて、それぞれの蛋白質のサブユニットが解離する変性条件下での MALDI-TOF 質量分析法による構成コンポーネントの同定を目的とした確認試験(アプロサイエンス社受託)を行なった。シナピン酸とウシ由来血清アルブミン(約66 kDa)の+1価から+5価のピークの質量電荷比(m/z)値をそれぞれイオン化用マトリックスならびに m/z ピーク値に関する外部標準とした解析の結果、各蛋白質分子中の結合に関与していないサブユニット由来のシグナル(ピーク m/z 値:Avi-PEG4-MTZ サブユニット、15895.5; hFasLECD-PEG3-TCOサブユニット、20698.9)に加えて、両蛋白質分子のサブユニットが TCO 基–MTZ 基間の付加反応により共有結合したコンポーネントに由来する構造に対応するシグナル(ピーク m/z 値:36854.0)を同定することができた[2]。
一方、細胞死誘導活性については、ヒト結腸がん由来の株化細胞の1種である HT-29 細胞を被検細胞として、架橋用の抗 FLAG 配列 M2 モノクローナル抗体(Sigma 社製)の存在および非存在条件下における FLAG-GlyCys(Gly)4-hFasLECD の N-エチルマレイミド修飾体に関する評価、ならびにヒトインターフェロンガンマ(IFN-γ)による前処理の存在および非存在条件下での FLAG-GlyCys(Gly)4-hFasLECD に関するフルオレセイン5-マレイミド修飾体および上記の hFasLECD-PEG3-TCO と Avi-PEG4-MTZ の1:1架橋体の2種類の試料に関する評価を MTT アッセイ法による細胞生存率測定試験(生体分子計測研究所社受託)により行なった[2]。いずれの場合にも hFasLECD に関する部位特異的化学修飾体試料のみの場合には有意な細胞死誘導活性は検出されなかった。この結果は生体内において膜タンパク質である hFasL からマトリックスメタロプロテアーゼの一種である MMP7 による切断により生成することが知られている可溶型の hFasLECD が非常に弱いアポトーシス誘導活性しか示さないという実験結果[76]と一致している。他方、上記のようなアポトーシス誘導を増強させるための処理を施した場合、前者では 2 μgmL-1 の架橋用抗体共存下で100または 1000 ngmL-1 の hFasLECD 試料を用いた72時間の処理により約50%への細胞生存率の低下が[68]、また後者については、100 IUmL-1 の IFN-γ による24時間の前処理存在条件下において、いずれの試料を使用した場合にも100または 1000 ngmL-1 の hFasLECD 化学修飾体による72時間の処理によりほぼ全ての被検細胞の死滅が観測された(図10)[2]。
上記の抗 FLAG 配列抗体を用いる架橋処理あるいは IFN-γ を用いる前処理を施した条件下における hFasLECD の各部位特異的化学修飾体による細胞死誘導後の被検細胞を光学顕微鏡により観察したところ、いずれの場合についてもアポトーシス誘導の結果生じたと考えられる擬円形に収縮した形態の細胞の特徴を示していた(図10)[1,2]。前者の場合、抗体による架橋により少なくとも2組以上の hFasLECD と hFasR の複合体が近接して形成される[29]。一方、後者に関しては、IFN-γ はヒト体内において hFasL と同様にナチュラルキラー(NK)細胞ならびに細胞障害性 T リンパ球(CTL)細胞から産生されるサイトカインとして標的細胞上の特異的受容体を介した生体防御活性の発現に関わっていることが知られており[77]、標的細胞表面上の hFasR 分子の増加や局在化を誘起することにより hFasL によるアポトーシス誘導に対する感受性を高めることにも寄与していると考えられている[28,78,79]。本研究で得られた実験結果は、可溶型の hFasLECD を用いた場合でも標的細胞表面に発現する FasR との複合体をある一定以上の密度で形成させることにより効率的なアポトーシスによる細胞死を誘導することが可能である、というこれまでの研究に基づく作業仮説[80]を改めて支持するものである。加えて、後者の系においては hFasL の主な供給源である NK 細胞や CTL 細胞から分泌生産される IFN-γ を利用していることから、前者のような元来ヒト体内には存在しない FLAG タグ配列を認識する架橋用抗体分子を用いる場合に比べて、より生体内で生じているアポトーシスに近い機序での細胞死の誘導がなされているのではないかと推定される。
4. 今後の展望
上述したように、hFasLECD の P. pastoris を宿主とする各種誘導体の発現生産ならびに発現産物に関する部位特異的化学修飾体の調製を手段とする高機能化を目指した分子デザインは、未だようやく基本的な方法論が構築されつつある段階である。2010年に日本インターフェロン・サイトカイン学会より出版された書籍「サイトカインハンティング」にも詳しく記述されているように、日常ヒトの免疫系を巧みに制御している各種の重要なサイトカイン分子の発見や同定には、日本人研究者が深く関わったものが多い[4]。とりわけ本稿で対象とした hFasL は、1.1項でも述べたように標的細胞に対するアポトーシスの誘導という医療応用上有用な特異な機能を有している[5]。そのため、発見当初から単にそのヒト体内での生理学的ならびに病理学的役割の面からの興味に留まらず、がんや自己免疫疾患など薬物による治療という観点からは未だ医療上の目標が充分に達成されたとは言い難い重大な疾病を対象とした臨床応用が強く意識されると共に、期待も高かったのではないかと思われる[81,82]。
しかしながら、初期に行われた実験用マウスを用いた全身性投与実験の際に生じた劇症肝炎の発症が主因と考えられる死亡事例[19,83]からも推察されるように、機能性蛋白質素子として hFasLECD 単体やアゴニスト型抗 hFasR モノクローナル抗体のみを用いた治療法の開発はかなり困難を伴うのではないかと考えられる。この課題を解決するための糸口の一つとして、これまでにもがん細胞等への特異的なターゲティングのための scFv 型抗体フラグメントとの遺伝子レベルでの結合を手段とした融合蛋白質の調製を始めとする hFasLECD に関する各種の誘導体の設計、開発が試みられてきた(表1)。本稿で紹介した部位特異的化学修飾法による融合蛋白質分子の調製は、原理的には scFv 型フラグメントだけでなく全長分子を含む他の各種の形態の抗体分子等にも適用可能ではないかと推測され、新規な形態の機能性素子成分を含む治療用融合蛋白質分子の創製につながると考えられる。また、hFasLECD はマウス由来の FasR に対しても特異的に結合し、アポトーシス誘導活性を示すことが知られている[84]。P. pastoris で生産された可溶型 hFasLECD は、マウス FasR を発現するリンパ球由来の W4 細胞に対する細胞障害活性に関する試験管内での試験においては当該レセプターに対するアゴニスト型モノクローナル抗体の一つである Jo2 に比べて少なくとも20倍以上の高い比活性を有することが示されており、またマウスを用いた肝臓への毒性に関する生体内での試験ではその障害活性の程度は Jo2 に比べて50分の1以下であることも明らかになっている[19,34]。マウスを用いたこれらの実験結果はヒトを対象とした細胞死誘導型治療用蛋白質の開発を目指す上でのアゴニスト型抗 hFasR モノクローナル抗体に対する hFasLECD の優位性を暗に示すものであるが、適切な部位特異的化学修飾の適用により正常組織への重篤な副作用がさらに軽減されることが望まれる。
アビジンとの架橋体は各種のビオチンを坦持した分子と強く結合すると考えられ、多様な機能性分子や材料と安定な複合体を形成させることも可能である。また、細胞表面に表出する hFasR 分子は IFN-γ 以外にもウイルスの感染[82]、臨床的に用いられている抗がん剤[85]、γ線照射[86]あるいは高濃度のグルコース[87]等の作用により有意に増加することが知られている。この現象は、3.5項で例示したように、単独では弱い可溶性 hFasLECD 誘導体の標的細胞に対する細胞死誘導活性を増強させる手段として実際の疾病治療法の開発においても応用できる可能性を秘めているように思われる。血清などの各種体液中に含まれる可溶型の hFasRECD は将来的ながんによる死亡[88]、糖尿病や心血管病の発症増加[89]あるいはある種の急性腎臓病の進展予後[90]などに関するリスク因子として、各種の疾病に関わる予測のためのバイオマーカーとしての有用性が指摘されている。また、hFasL に結合するもう一つの特異的レセプターであり、体内でのがんの増殖や炎症反応に関する有用なバイオマーカーとしても知られている hDcR3[91]についても臨床上の固形がん患者の予後の推定に利用できることが示唆されている[92]。hFasRECD や hDcR3 のみを選択的に認識できる hFasLECD の部位特異的化学修飾体は疾病の診断用システムや治療薬の開発のための評価用システムを構築する際の分子ツールとしての利用も可能ではないだろうか。
P. pastoris を宿主とする発現生産系は、これまでに多種類の異種組換え蛋白質に関する大量調製の実績が豊富にあり高い確率で哺乳動物由来の培養細胞を用いる発現系に比べてより短期間かつ低コストでの目的蛋白質の生産が可能という、産業応用上の利点を有している[13,15]。また、哺乳動物細胞を用いる発現生産の場合のようなヒトに感染する可能性のある病原性ウイルスの混入についても危険性は少ないものと思われる。他方、付加される糖鎖構造が高マンノース型であり哺乳動物細胞由来のものとは異なることから、この糖鎖構造の違いに起因する免疫原性の惹起などの副作用が心配されるが、近年では哺乳動物細胞由来のものと類似した複合型糖鎖の付加が可能な株の開発も進んできた[93]。さらに現在では、知的財産面での制約についても、ATUM 社から野生型株に加えて PEP4 や PRB1 などの液胞プロテアーゼに関する欠損株を含む発現生産に適した数種類の “IP-Free Pichia strains” やそれらを用いる蛋白質の生産に適した発現ベクターが市販されており(コスモバイオ社扱い)、これらの製品を利用して製造した蛋白質の商用目的での使用についてはライセンスの取得を必要としないようである[94]。
近年、蛋白質分子の部位特異的化学修飾に適した生体直交型(bioorthogonal)の化学反応が種々開発されてきている[95]。中でも、本研究において使用した MTZ 基を含むジエン分子と TCO 基を含むジエノフィル分子との間での iEDDA 付加反応は、非常に高い反応速度定数(10~102 M-1s-1)を有し、かつ各官能基が通常蛋白質分子が変性を起こしにくい生理的 pH の緩衝液条件下において適度な化学的安定性を兼ね備えているため、とりわけ蛋白質分子間の架橋反応として大変利用価値が高いと考えられる[96,97]。一方、高分子量の蛋白質分子間の部位特異的化学架橋体を調製する場合、目的とする架橋体を高収量で経済的に得るためには反応条件に関する予備的検討が重要と思われる。最終的に精密な糖鎖構造を考慮に入れた哺乳動物細胞発現系で生産した被化学修飾用蛋白質が求められる場合においても、その前段階としての条件探索的研究では試験に供するための精製試料は多量に必要になると予想される。この点でも P. pastoris を宿主とする発現生産実験は比較的安価に実施可能であるため有用であろう。本稿で紹介した hFasLECD 誘導体の P. pastoris を宿主とした発現生産とその部位特異的化学修飾体の調製が、近い将来に実際の医療面での課題解決を図る上での有効な手段に発展することを期待したい。
5. 謝辞
蛋白質分子の立体構造図は日本蛋白質構造データバンク(PDBj)に登録されている座標データならびに PDBj から提供されているグラフィクス描画プログラム jV を用いて、また化学構造式は ACD 社のソフトウェア ChemSketch(Freeware)2016.1.1 を使用して作成しました。ここに記して謝意を表します。最後に、本総説中に記載した研究の遂行に当り多大な御協力を賜りました研究支援関連企業の担当者の方々、本稿の執筆を御奨励いただくと共に発表に際して大変お世話になりました産業技術総合研究所・バイオメディカル研究部門長、近江谷克裕博士ならびに同部門蛋白質デザイン研究グループ長、広田潔憲博士を始めとする関係者の皆様に深く感謝致します。
6. 引用文献
- Muraki, M., AIMS Bioengineering, 5, 39–62 (2018)
- Muraki, M. & Hirota, K., BMC Res. Notes, 11, 395 (2018)
- FASLG Fas ligand [Homo sapiens (human)]
- 長田 重一、米原 伸, サイトカインハンティング, 第8章 G-CSF, Fas, 253–67, 日本インターフェロン・サイトカイン学会編, 京都大学学術出版会 (2010)
- 須田 貴司, シリーズ・バイオサイエンスの新世紀 第6巻 細胞の誕生と死, 第3章 細胞死, ② FasとFasリガンド, 143–59, 長田 重一、山本 雅/日本生化学会編, 共立出版 (2001)
- Wajant, H. ed., Medical Intelligence Unit, Fas Signaling, Landes Bioscience, Georgetown / Springer Science+Business Media, New York (2006)
- Yi, F. et al., Trend. Mol. Med., 24, 642–53 (2018)
- Bodmer, LC. et al., Trends Biochem. Sci., 27, 19–26 (2002)
- Pitti, RM. et al., Nature, 396, 699–703 (1998)
- Liu, W. et al., Structure, 24, 2016–23 (2016)
- Ahmad, M. et al., Appl. Microbiol. Biotechnol., 98, 5301–17 (2014)
- Prabhu, AA. et al., Curr. Pharma. Biotechnol., 18, 1200–23 (2017)
- Pichia protocols (ed. Higgins DR. & Cregg JM.), Methods in Molecular Biology, 103 (Ser. Ed. Walker, JM), Humana Press, Totowa (1998)
- Pichia protocols Second Edition (ed. Cregg JM.), Methods in Molecular Biology, 389 (Ser. Ed. Walker, JM), Humana Press, Totowa (2007)
- 内藤 忠相、杉山 賢司、目的別で選べるタンパク質発現プロトコール、第3章 3.酵母、③タンパク質の発現、162–71, 永田 恭介、奥脇 暢 編、羊土社 (2010)
- 櫻井 一正、蛋白質科学会アーカイブ、1, e018 (2008)
- 村木 三智郎、蛋白質科学会アーカイブ、7, e078 (2014)
- Muraki, M., Prot. Pept. Lett., 19, 867–79 (2012)
- Tanaka, M. et al., J. Immunol., 158, 2303–09 (1997)
- Muraki, M., Prot. Expr. Purif., 50, 137–46 (2006)
- Luo, Z. et al., Biochem. Eng. J., 62, 86–91 (2012)
- Sun, KH. et al., Enzyme Microb. Technol., 36, 527–34 (2005)
- 萩原 義久、蛋白質科学会アーカイブ、1, e001 (2008)
- 浜田 寛之、松岡 常吉、蛋白質科学会アーカイブ, 1, e035 (2008)
- 梅津 光央、蛋白質科学会アーカイブ, 1, e038 (2008)
- Liu, W. et al., Mol. Biotechnol., 57, 914–22 (2015)
- 柴田 俊生 他、蛋白質科学会アーカイブ, 2, e052 (2009)
- Mollinedo, F. & Gajate, C., Drug Resist. Updates, 9, 51–73 (2006)
- Holler, N. et al., Mol. Cell. Biol., 23, 1428–40 (2003)
- Eisele, G. et al., Neuro Oncol., 13, 155–64 (2011)
- Eisele, G. et al., Anticancer Res., 33, 3563–71 (2013)
- http://www.oncologyventure.com/pipeline/apo010/
- Daburon, S. et al., Plos One, 8, e54000 (2013)
- Timmer, T. et al., J. Pathol., 196, 125–34 (2002)
- 橋口 隆生 他、蛋白質科学会アーカイブ、1, e017 (2008)
- 浅野 竜太郎, 蛋白質科学会アーカイブ, 2, e050 (2009)
- Samel, D. et al., J. Biol. Chem., 278, 32077–82 (2003)
- Bremer, E. et al., Cancer Res., 68, 597–604 (2008)
- Bremer, E. et al., Blood, 107, 2863–70 (2006)
- Chien, MH. et al., Mol. Cancer Ther., 16, 1102–13 (2017)
- Chan, DV. et al., J. Biomed. Sci., 20, 16 (2013)
- Hemmerle, T. et al., J. Biotechnol., 172, 73–6 (2014)
- Orbach, A. et al., Am. J. Pathol., 177, 3159–68 (2010)
- Aronin, A. et al., J. Hematol. Oncol., 7, 64 (2014)
- Zhang, W. et al., Mol. Immunol., 50, 150–9 (2012)
- Shi, W. et al., Graefe’s Arch. Clin. Exp. Ophthalmol., 245, 1691–7 (2007)
- Feng, YG. et al., Transplant. Proc., 37, 2379–81 (2005)
- Kitano, H. et al., J. Gene. Med., 14, 642–50 (2012)
- Morello, A. et al., Plos One, 8, e73375 (2013)
- Franke, DDH. et al., Mol. Immunol., 44, 2884–92 (2007)
- Cereghino, JL. & Cregg, JM., FEMS Microbiol. Rev., 24, 45–66 (2000)
- Jigami, Y. et al., Gene, 43, 273–9 (1986)
- Nagahora, H. et al., Eur. J. Biochem., 210, 989–97 (1992)
- Ikemura, T., Mol. Biol. Evol., 2, 13–34 (1985)
- Jacobs, PP. et al., Nat. Protoc., 4, 58–70 (2009)
- Helenius, A. & Aebi, M., Annu. Rev. Biochem., 73, 1019–49 (2004)
- Pfeffer, M. et al., Appl. Microbiol. Biotechnol., 93, 2503–12 (2012)
- Muraki, M., Prot. Expr. Purif., 60, 205–13 (2008)
- Orlinick, JR. et al., J. Biol. Chem., 272, 32221–9 (1997)
- Gasteiger, E. et al., Protein Identification and Analysis Tools on the ExPAsy Server (ed Walker JM.) 571–607, Humana Press (2005)
- Muraki, M. & Hirota, K., BMC Biotechnol., 17, 56 (2017)
- Mahiou, J. et al., Biochem. J., 330, 1051–8 (1998)
- 久武 幸司、福田 綾、目的別で選べるタンパク質発現プロトコール、第3章 2.昆虫細胞、120–44, 永田 恭介、奥脇 暢 編、羊土社 (2010)
- 前川 憲一、蛋白質科学会アーカイブ, 1, e022 (2008)
- 本庄 栄二郎、 蛋白質科学会アーカイブ, 1, e029 (2008)
- Muraki, M. & Honda, S., Prot. Expr. Purif., 73, 209–16 (2010)
- Muraki, M & Honda, S., Prot. Expr. Purif., 80, 102–9 (2011)
- Muraki, M., BMC Biotechnol., 14:19 (2014)
- Couderc, R. & Baratti, J., Agric. Biol. Chem., 44, 2279–89 (1980)
- Banner, DW. Et al., Cell, 73, 431–45 (1993)
- Hermanson, GT., Bioconjugate Techniques (Third Edition), Academic Press (2013)
- Shafer, DE. Et al., Anal. Biochem., 282, 161–4 (2000)
- Muraki, M., SpringerPlus, 5:997 (2016)
- https://www.lumiprobe.com/protocols/nhs-ester-labeling.pdf
- 石川 榮治, 生物化学実験法 27 酵素標識法, 第Ⅲ章, 31–50, 瓜谷 郁三 他編、学会出版センター (1991)
- Schneider, P. et al., J. Exp. Med., 187, 1205–13 (1998)
- Schroder, K. et al., J. Leukoc. Biol., 75, 163–89 (2004)
- Xu, X. et al., Cancer Res., 58, 2832–37 (1998)
- Schwartzberg, LS. et al., Clin. Cancer Res., 8, 2488–98 (2002)
- Thorburn, A., Cell. Signal., 16, 139–44 (2004)
- Nagata, S., Cell, 88, 355–65 (1997)
- 中西 義信, 蛋白質 核酸 酵素, 41, 2552–8 (1996)
- Ogasawara, J. et al., Nature, 364, 806–9 (1993)
- Takahashi, T. et al., Int. Immunol., 6, 1567–74 (1994)
- Micheau, O. et al., J. Natl. Cancer Inst., 89, 783–9 (1997)
- Reap, EA. et al., Proc. Natl. Acad. Sci. USA, 94, 5750–5 (1997)
- Maedler, K. et al., Diabetes, 50, 1683–90 (2001)
- Tamakoshi, A. et al., Int. J. Cancer, 123, 1913–6 (2008)
- Mattisson, IY. et al., EBioMedicine, 26, 187–97 (2017)
- Bhatraju, PK. et al., Crit. Care, 21, 217 (2017)
- Hsieh, SL. & Lin, WW., J. Biomed. Sci., 24, 39 (2017)
- Ge, H. et al., Clin. Chim. Acta, 481, 126–31 (2018)
- Liu, L. et al., Biologicals, 39, 205–10 (2011)
- 1.5. License for Pichia Strains, ATUM 社 TERMS & CONDITIONS
- Oliveira, BL., Chem. Soc. Rev., 46, 4895–950 (2017)
- Wu, H. & Devaraj, NK., Top. Curr. Chem., 374, 3 (2016)
- Mayer, S. & Lang, K., Synthesis-Stuttgart, 49, 830–48 (2017)
-
![図1: 野生型ヒトFasリガンドを構成するドメイン構造(模式図)略号:MEM, 膜貫通ドメイン; TRI, サブユニット3量体構造を形成するために必要な領域; TNFHD, 他のTNFリガンドスーパーファミリーに属する蛋白質と配列相同性を有する領域。各数字ならびに矢印はそれぞれN末端残基を1とするアミノ酸残基の番号ならびにN-結合型糖鎖付加サイトを構成するAsn残基の位置を示す。NH2, N末端;COOH, C末端。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]](/archives/images/pichia_03/pichia_03_Fig_01.png)
図1: 野生型ヒト Fas リガンドを構成するドメイン構造(模式図)略号:MEM, 膜貫通ドメイン; TRI, サブユニット3量体構造を形成するために必要な領域; TNFHD, 他の TNF リガンドスーパーファミリーに属する蛋白質と配列相同性を有する領域。各数字ならびに矢印はそれぞれ N 末端残基を1とするアミノ酸残基の番号ならびに N-結合型糖鎖付加サイトを構成する Asn 残基の位置を示す。NH2, N 末端;COOH, C 末端。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1] -
![図2: 野生型ヒト Fas リガンド細胞外ドメイン(hFasLECD)の一次構造と二次構造。a) 一次構造。各アミノ酸残基を1文字表記し、X線結晶解析[10]で明らかにされたβシート構造の位置をアミノ酸配列下部の青色バーで示した。また、本文中で触れた代表的なアミノ酸残基に残基番号を記した。b) 二次構造(リボン表記)。この立体構造モデルにおけるN末端残基(L143)ならびにC末端残基(L281)の位置を示した。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_02.png)
図2: 野生型ヒト Fas リガンド細胞外ドメイン(hFasLECD)の一次構造と二次構造。a) 一次構造。各アミノ酸残基を1文字表記し、X 線結晶解析[10]で明らかにされたβシート構造の位置をアミノ酸配列下部の青色バーで示した。また、本文中で触れた代表的なアミノ酸残基に残基番号を記した。b) 二次構造(リボン表記)。この立体構造モデルにおけるN末端残基(L143)ならびにC末端残基(L281)の位置を示した。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。 -
![図3: hFasLECD-hDcR3複合体の立体構造(PDB ID: 4MSV [10])。野生型hFasLECDとhDcR3の複合体に関する立体構造。hFasLECDのN末端アミノ酸残基(この立体構造モデルにおいてはLeu143)、当該ドメイン中に存在する3箇所のN結合型糖鎖付加部位を構成するAsn残基(Asn184, Asn250およびAsn260)ならびに1箇所の分子内ジスルフィド結合を形成する一対のシステイン残基(Cys202およびCys233)については色を変えて示した。配色:シアン、hFasLECD;オレンジ、N末端アミノ酸残基(このモデルではLeu143残基);赤、N結合型糖鎖付加部位を構成するAsn残基;黄、Cys残基;白、hDcR3。各N結合型糖鎖付加部位ならびにジスルフィド結合の位置を矢印で示した。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_03.png)
図3: hFasLECD-hDcR3 複合体の立体構造(PDB ID: 4MSV [10])。野生型 hFasLECD と hDcR3 の複合体に関する立体構造。hFasLECD の N 末端アミノ酸残基(この立体構造モデルにおいては Leu143)、当該ドメイン中に存在する3箇所の N 結合型糖鎖付加部位を構成する Asn 残基(Asn184, Asn250 および Asn260)ならびに1箇所の分子内ジスルフィド結合を形成する一対のシステイン残基(Cys202 および Cys233)については色を変えて示した。配色:シアン、hFasLECD;オレンジ、N末端アミノ酸残基(このモデルでは Leu143 残基);赤、N 結合型糖鎖付加部位を構成する Asn 残基;黄、Cys 残基;白、hDcR3。各 N 結合型糖鎖付加部位ならびにジスルフィド結合の位置を矢印で示した。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。 -
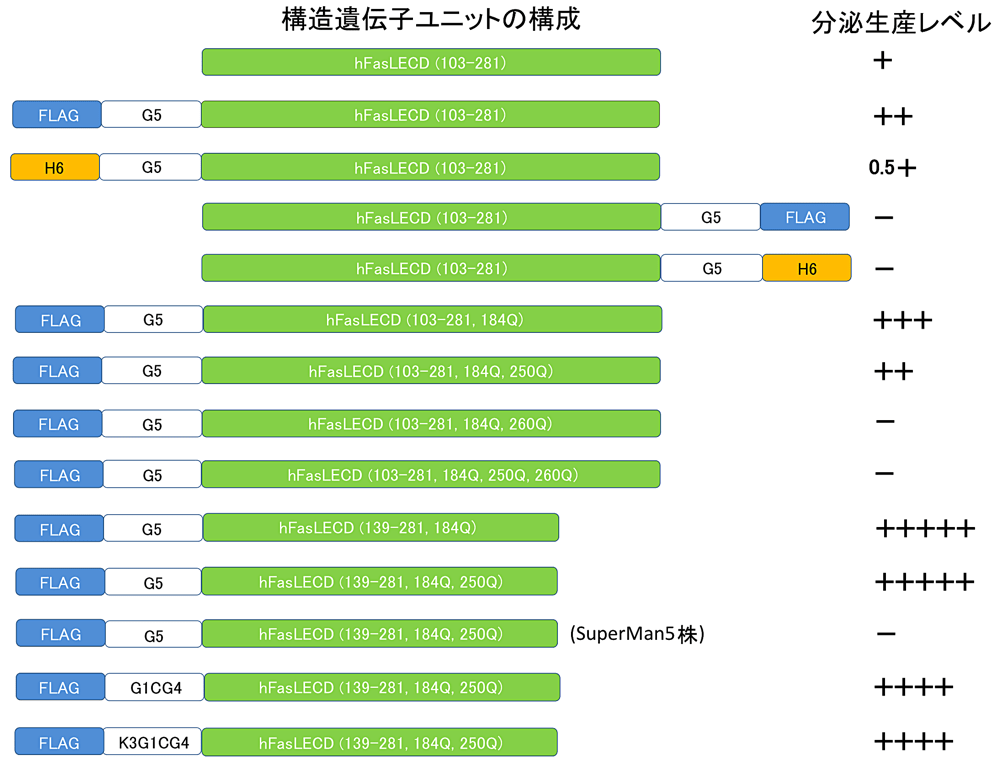
図4: P. pastoris を宿主とした hFasLECD 誘導体の発現における構造遺伝子ユニットの構成の違いが分泌生産レベルにもたらす影響。略号:FLAG, AspTyrLysAspAspAspAspLys; H6, (His)6; G5, (Gly)5; G1CG4, GlyCys(Gly)4; K3G1CG4, (Lys)3GlyCys(Gly)4。株名を記載していないものは全て GS115 株を用いた結果を示す。培養上清の SDS-PAGE 分析における各発現産物に対応するバンドの濃さから推定された、それぞれの構造遺伝子ユニットを用いた場合のおよその相対的な分泌生産レベルを+の数で表した。-は検出が困難であることを示す。 -
![図5: ディスポーザブルカルチャーバッグシステムおよび培養上清濃縮用限外ろ過システム。a, b) ディスポーザブルカルチャーバッグシステム(a, 模式図、b, 実物写真)。ディスポーザブルカルチャーバッグ、微生物培養用シングルユースバッグCB5-1/CB10-1 (藤森工業社製)。c) 培養上清濃縮用限外ろ過システム(模式図)。限外ろ過デバイス、オメガメンブレンミニメイトTFFカプセルOA010C12(ポール社製);3連ペリスタポンプ、チューブポンプISO-313T(アズワン社製)。配管用チューブとしてはペリスタポンプを経由する部分はファーメドチューブ(サンゴバン社製)を、それ以外はタイゴンチューブまたはシリコンチューブを使用。限外ろ過デバイスの濃縮液側出口近傍に設置したスクリュークランプを用いた加圧により、ろ液の排出速度を調節。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_05.jpg)
図5: ディスポーザブルカルチャーバッグシステムおよび培養上清濃縮用限外ろ過システム。a, b) ディスポーザブルカルチャーバッグシステム(a, 模式図、b, 実物写真)。ディスポーザブルカルチャーバッグ、微生物培養用シングルユースバッグ CB5-1/CB10-1 (藤森工業社製)。c) 培養上清濃縮用限外ろ過システム(模式図)。限外ろ過デバイス、オメガメンブレンミニメイト TFF カプセル OA010C12(ポール社製);3連ペリスタポンプ、チューブポンプ ISO-313T(アズワン社製)。配管用チューブとしてはペリスタポンプを経由する部分はファーメドチューブ(サンゴバン社製)を、それ以外はタイゴンチューブまたはシリコンチューブを使用。限外ろ過デバイスの濃縮液側出口近傍に設置したスクリュークランプを用いた加圧により、ろ液の排出速度を調節。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。 -
![図6: hFasLECD部位特異的化学修飾体の調製に使用した反応。a) hFasLECD(139-281, Q184, Q250)誘導体に接続されたN末端タグ配列中のCys残基とマレイミド基を含む分子1との間のチオール-エン付加反応、b) TCO基を含む分子2とMTZ基を含む分子3との間のiEDDA付加反応。分子2または分子3のいずれかにhFasLECD誘導体が含まれる。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[61]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_06.png)
図6: hFasLECD 部位特異的化学修飾体の調製に使用した反応。a) hFasLECD(139-281, Q184, Q250)誘導体に接続された N 末端タグ配列中の Cys 残基とマレイミド基を含む分子1との間のチオール-エン付加反応、b) TCO 基を含む分子2と MTZ 基を含む分子3との間の iEDDA 付加反応。分子2または分子3のいずれかに hFasLECD 誘導体が含まれる。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[61]を改変して作成。 -
![図7: hFasLECDの誘導体化ならびに他の機能性蛋白質分子との結合後のクエンチング反応に使用したTCO基またはMTZ基を含有する試薬の化学構造。配色:青、TCO基;赤、MTZ基。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_07.png)
図7: hFasLECD の誘導体化ならびに他の機能性蛋白質分子との結合後のクエンチング反応に使用した TCO 基または MTZ 基を含有する試薬の化学構造。配色:青、TCO 基;赤、MTZ 基。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。 -
![図8: hFasLECD、Avi、rFabの立体構造とhFasLECD-Aviの構造模式図。a)立体構造、各蛋白質を構成する原子の座標データ(PDB ID: hFasLECD、4MSVより抜粋; Avi、1AVE; rFab、4HBC)を用いて描画した。構成原子種別(灰色、炭素;赤色、酸素;青色、窒素;黄色、硫黄)に配色。b)hFasLECD-Avi 1:1架橋体分子の構造模式図、青線、TCO-PEG3スペーサー部位;赤線、MTZ-PEG4スペーサー部位;青丸、クエンチング反応により付加されたTCO-NH2分子;黒丸、TCO基とMTZ基の付加結合部位。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_08.png)
図8: hFasLECD、Avi、rFab の立体構造と hFasLECD-Avi の構造模式図。a)立体構造、各蛋白質を構成する原子の座標データ(PDB ID: hFasLECD、4MSV より抜粋; Avi、1AVE; rFab、4HBC)を用いて描画した。構成原子種別(灰色、炭素;赤色、酸素;青色、窒素;黄色、硫黄)に配色。b)hFasLECD-Avi 1:1架橋体分子の構造模式図、青線、TCO-PEG3 スペーサー部位;赤線、MTZ-PEG4 スペーサー部位;青丸、クエンチング反応により付加された TCO-NH2 分子;黒丸、TCO 基と MTZ 基の付加結合部位。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。 -
![図9: hFasLECD-PEG3-TCOと他の機能性蛋白質のMTZ誘導体の架橋反応混合物の分析。図中に記載した各混合モル比のhFasLECD-PEG3-TCO(TCO)と他の機能性蛋白質のMTZ誘導体(MTZ)の反応混合物のサイズ排除クロマトグラフィーにおけるプロフィールと各ピークの溶出時間を示した。a) Avi-PEG4-MTZとの反応混合物(28℃、1h)、ピークラベル: 0, 非架橋体; *, 1:1架橋体、b) rFab’-PEG4-MTZとの反応混合物(25℃、1h)、ピークラベル: 0, 非架橋体; 1, 1:1架橋体; 2, 1:2架橋体; 3, 1:3架橋体。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[61]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_09.png)
図9: hFasLECD-PEG3-TCO と他の機能性蛋白質の MTZ 誘導体の架橋反応混合物の分析。図中に記載した各混合モル比の hFasLECD-PEG3-TCO(TCO)と他の機能性蛋白質の MTZ 誘導体(MTZ)の反応混合物のサイズ排除クロマトグラフィーにおけるプロフィールと各ピークの溶出時間を示した。a) Avi-PEG4-MTZ との反応混合物(28℃、1h)、ピークラベル: 0, 非架橋体; *, 1:1架橋体、b) rFab’-PEG4-MTZ との反応混合物(25℃、1h)、ピークラベル: 0, 非架橋体; 1, 1:1架橋体; 2, 1:2架橋体; 3, 1:3架橋体。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[61]を改変して作成。 -
![図10: hFasLECD-Avi架橋体による細胞死誘導におけるIFN-γ前処理の効果。a) MTTアッセイ。被検細胞:ヒト結腸がん由来HT-29細胞。横軸に示す各濃度のヒトIFN-γによる24時間の前処理に引き続く各被検試料による72時間処理後の細胞生存率を、PBS緩衝液のみによる場合の平均値を100として示した。被検試料:青、PBS緩衝液のみ;緑、フルオレセイン担持ウシ血清由来アルブミン(ネガティブコントロール);オレンジ、hFasLECD-Avi 1:1架橋体。被検試料濃度:左、100 ng/ml; 右、1000 ng/ml。各条件についての4回の独立した測定より平均値ならびに誤差を算出した。 b) HT-29細胞の形態写真。各試薬(左、PBS緩衝液のみ; 右、100 IU/ml ヒトIFN-γを含むPBS緩衝液)による24時間の前処理後にhFasLECD-Avi 1:1架橋体試料(100 ng/ml)による72時間の処理を実施したもの。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[2]を改変して作成。](/archives/images/pichia_03/pichia_03_Fig_10.jpg)
図10: hFasLECD-Avi 架橋体による細胞死誘導における IFN-γ 前処理の効果。a) MTT アッセイ。被検細胞:ヒト結腸がん由来 HT-29 細胞。横軸に示す各濃度のヒト IFN-γ による24時間の前処理に引き続く各被検試料による72時間処理後の細胞生存率を、PBS 緩衝液のみによる場合の平均値を100として示した。被検試料:青、PBS 緩衝液のみ;緑、フルオレセイン担持ウシ血清由来アルブミン(ネガティブコントロール);オレンジ、hFasLECD-Avi 1:1架橋体。被検試料濃度:左、100 ng/ml; 右、1000 ng/ml。各条件についての4回の独立した測定より平均値ならびに誤差を算出した。 b) HT-29 細胞の形態写真。各試薬(左、PBS 緩衝液のみ; 右、100 IU/ml ヒト IFN-γ を含む PBS 緩衝液)による24時間の前処理後に hFasLECD-Avi 1:1架橋体試料(100 ng/ml)による72時間の処理を実施したもの。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[2]を改変して作成。 -
![表1: 医療応用を目的として遺伝子結合法により調製されたヒトFasリガンド細胞外ドメイン融合蛋白質。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。](/archives/images/pichia_03/pichia_03_Table_01.png)
表1: 医療応用を目的として遺伝子結合法により調製されたヒト Fas リガンド細胞外ドメイン融合蛋白質。©Michiro Muraki((クリエイティブ・コモンズ・ライセンス(表示4.0国際))[1]を改変して作成。
概要
ヒト Fas リガンドはがんなどの重大な疾病の原因となる細胞に対して細胞死を誘導する能力を有する膜蛋白質であり、その細胞外ドメインは標的細胞上のFasレセプターに特異的に結合しアポトーシス実行のためのシグナル伝達の引き金を引く役割を担っている。この総説では、ヒト Fas リガンド細胞外ドメインを対象とした Pichia pastoris 酵母を宿主とする各種誘導体の発現生産系の構築および部位特異的化学修飾体の調製を手段とする医療応用上有用な機能を持つ蛋白質分子の創製を目指した分子デザインのための方法の開発に焦点を当てて紹介する。本総説では、関連する周辺の研究の進展ならびにこれまでに発表した総説や原著論文において充分に詳しく記述できなかった事項や一部未発表のデータも含めて、経緯と現状を省みると共に、今後の研究の発展への期待を含めた展望についても触れる。尚、本稿は著者が2018年1月に発表した英文総説[1]に加筆と修正による改訂を加えたものを基に、その後に発表した原著論文[2]の内容を追補して作成したものである。
目次
- はじめに
- 1.1 ヒト Fas リガンドとは
- 1.2 Pichia pastoris を宿主とする異種蛋白質の発現生産系
- 1.3 これまでに開発されてきたヒト Fas リガンド細胞外ドメイン(hFasLECD)の異種発現生産系
- 1.4 これまでに開発されてきた医療応用を目的とした hFasLECD 誘導体
- hFasLECD 誘導体の Pichia pastoris 分泌発現生産系を用いた調製
- 2.1 分泌発現生産系の構築
- 2.2 タグ配列付加ならびに糖鎖付加部位欠失の影響
- 2.3 N 末端領域の除去が分泌生産量に与える効果
- 2.4 陽イオン交換クロマトグラフィーによる精製
- 2.5 ヒト Fas レセプター細胞外ドメインに対する結合活性の確認
- 2.6 ディスポーザブルカルチャーバッグを用いた培養システムの開発
- hFasLECD の部位特異的化学修飾体の調製とキャラクタリゼーション
- 3.1 N 末端タグ配列中にシステイン残基を有する誘導体の設計と発現生産
- 3.2 部位特異的化学修飾体の調製法の検討
- 3.3 低分子性蛍光色素による化学修飾体の調製
- 3.4 他の機能性蛋白質分子との架橋体の調製
- 3.5 共有結合構造の同定と細胞死誘導活性の評価
- 今後の展望
- 謝辞
- 引用文献
1. はじめに
1.1 ヒト Fas リガンドとは
ヒト Fas リガンド(hFasL)は、ヒトゲノムの第1番染色体上にコードされ[3]、腫瘍壊死因子(tumor necrosis factor/TNF)レセプタースーパーファミリーと名付けられた蛋白質グループの一員であるヒト Fas レセプター(hFasR/CD95/APO-1)を表出する標的細胞に対してアポトーシスによる死の引き金をひく、一回膜貫通型の糖蛋白質である。hFasR に代表される、いわゆるデスレセプターと結合して標的細胞の細胞死を誘導する性質のある蛋白質は一般的にデスリガンドと呼ばれている。FasL(CD95L/CD178/APO-1L)は TNFα、リンフォトキシンα(TNFβ)、TRAIL(CD253/APO-2L)、TWEAK(CD255/APO-3L)などと並んで代表的なデスリガンドの一つである。これらの蛋白質群は TNF リガンドスーパーファミリーと呼ばれ、標的細胞に対するアポトーシスによる死の誘導という注目すべき生物活性のために、その発見、同定以来、がんや関節リウマチ、全身性エリテマトーデスに代表される各種の自己免疫疾患などの重大な疾病の治療への貢献を目的とした数多くの研究がこれまでになされてきた。
FasL-FasR 系によるアポトーシス実行の特徴ならびにその医療応用の可能性を対象とした優れた和文の総説や解説についてはこれまでに多数発表されてきている。それらの中で最も解りやすくかつ包括的なものの一つに、Fas 抗原の発見者の一人でありその命名者でもある米原伸博士らと共に FasR の cDNA のクローニングによる同定[4]を行われた長田重一博士が共同の編集委員を務められた日本生化学会編の「シリーズ・バイオサイエンスの新世紀 第6巻 細胞の誕生と死」の中に含まれている、FasL 遺伝子のクローニングに初めて成功された須田貴司博士著の「第3章 細胞死:②FasとFasリガンド」が挙げられる[5]。また、英文の総説集として代表的なものの一つに、Herald Wajant 博士編の “Fas Signaling”[6]がある。FasL の発見と遺伝子のクローニングに関する経緯、基本的なシグナル伝達経路ならびにアポトーシスの誘導に関わる生体内での主要な生理的役割や病理的機能についてはこれらの著書にほぼ網羅されている。さらに、FasL-FasR 系を介するシグナル伝達経路は標的細胞の状態に応じて、アポトーシスだけではなくネクロプトーシスや細胞分化の誘導などにも関与することが指摘されており[6]、近年ではそれらの面からの自己免疫疾患や悪性腫瘍に対する免疫学的治療の可能性についても積極的に検討されるようになってきた[7]。
図1に hFasL 蛋白質を構成する全体的なドメイン構造を、また図2にその中で細胞外ドメイン部分の一次構造ならびに二次構造を示した。ヒト FasL 細胞外ドメイン(hFasLECD)については、以前から生理的条件下において同一サブユニットからなるホモ3量体構造を形成し、高さ約 6 nm の頂上部が欠損したベル型のピラミッド状の立体構造を有することが予測されていたが[8]、具体的な形状については、近年まで上記の「第3章 細胞死:②FasとFasリガンド」中に掲載されているコンピュータを用いた予想に基づいて作成された「Fas-Fas リガンド複合体の立体構造モデル」などが主なよりどころであった。その後2013年になり、X 線結晶構造解析を用いた詳細な立体構造に関する情報が米国アルバートアインシュタイン医科大学の Weifeng Liu 博士を筆頭とする Steven C. Almo 博士らのグループにより、現在のところ hFasL がヒトの体内において hFasR 以外に唯一特異的に結合すると考えられているデコイ(おとり)型レセプターであるヒト DcR3(hDcR3)[9]との複合体の形で蛋白質構造データバンク(PDB)に PDB ID:4MSV(公開日:2013-11-27)として登録された[10]。しかしながら、未だ野生型 hFasLECD 単体ならびにそれに対するアポトーシスシグナル伝達実行型受容体の細胞外ドメインであるヒト FasR 細胞外ドメイン(hFasRECD)との複合体の立体構造は明らかにされていない。そのため、野生型 hFasLECD の複合体形成過程における構造変化や複合体形成時の hFasRECD の場合と hDcR3 の場合の詳細な相互作用様式の相違の解明等については今後の検討課題として残されている。一方、hFasLECD 中のアミノ酸残基番号164-169に対応する AspThrTyrGlyIleVal の配列が同じく TNF リガンドスーパーファミリーに属する蛋白質分子の一つである TL1A 中の対応する配列である HisGluLeuGlyLeuAla に置換された変異体についての単体ならびにその hDcR3 との複合体については既に PDB に登録されている(PDB ID:5L19および5L36[10])(図3)。X 線結晶解析で明らかにされた立体構造によると、後述するアミノ酸残基番号139-281の領域は主に逆並行型のβ-シート構造からなる二次構造が発達しており(図2)、多くの水素結合を介した相互作用に裏打ちされたゆらぎの少ない熱力学的に安定な構造であることが示唆される。この二次構造形成領域は2002年に TNF リガンドスーパーファミリーに属する他の蛋白質との配列相同性から予測されたもの[8]と、よく一致していた。
hFasL 蛋白質分子を構成する各領域のうち、細胞外ドメインは標的細胞の表面に存在する hFasR に特異的に結合することでアポトーシス実行のためのシグナル伝達の引き金を引く役割を担っている。本稿では、この細胞外ドメインに関する各種誘導体の Pichia pastoris を宿主とする分泌発現生産系の構築ならびに部位特異的化学修飾を手段とした他の機能性分子との結合体の調製法の開発に焦点を当てて記述するが、本論に入る前にまず、P. pastoris を宿主とする異種蛋白質の発現生産系、これまでに開発されてきた hFasLECD の異種発現生産系ならびに将来的な医療分野での応用を目的として開発されてきた hFasLECD 誘導体について概略的に紹介する。
1.2 Pichia pastoris を宿主とする異種蛋白質の発現生産系
メタノール資化性酵母の一種であるピキア酵母(Pichia pastoris)は、単細胞性の微生物であることから蛋白質の発現生産実験を行う上で大腸菌と同様な取扱いの簡便さを有するだけでなく、同時に真核生物でもあるため哺乳動物細胞の場合と類似した品質管理機構を伴う高度な分泌生産能力を有している。そのため、ヒト由来のものを含む異種蛋白質の発現生産において多用され、近年ではゲノム編集を始めとする各種の合成生物学的手法による宿主としての改良も盛んに行なわれている[11,12]。一般的な異種組換え蛋白質生産用宿主としての P. pastoris の優位性や特徴についてはこれまでに発表された多くの総説やモノグラフ等において触れられているので、ここでは筆者が研究を進める過程でしばしば参照し、具体的な実験を行なうに当たって大変有用であった詳細なプロトコールが記載されている成書や技術資料について紹介するにとどめる。
P. pastoris を使用した異種蛋白質の発現生産系の開発に関する代表的な英文プロトコール集としては、いずれも Methods in Molecular Biology シリーズの一つとして Humana Press より刊行されている、David R. Higgins 博士と James M Cregg 博士により編集された “Pichia protocols”(1998年)[13]ならびに、その続編の “Pichia protocols Second Edition”(2007年)[14]が挙げられる。また P. pastoris を使用した発現生産実験に関する詳細な記述がされている和文のプロトコールの例としては羊土社より刊行されている実験医学別冊「目的別で選べる蛋白質発現プロトコール」(2010年)の第3章「3. 酵母、③タンパク質の発現」の項[15]に含まれている、内藤忠相博士と杉山賢司博士により著された「2. ピキア酵母におけるメタノールを用いたタンパク質発現-小スケールでの発現チェックおよび培養条件の至適化」ならびに「3. ピキア酵母におけるメタノールを用いたタンパク質発現-大スケール」の2つの解説記事、加えて日本蛋白質科学会編のオンライン版プロトコール集である「蛋白質科学会アーカイブ」中の「蛋白質の調製」のセクションに含まれている櫻井一正博士による「#018 メタノール資化性酵母 Pichia pastoris を用いた組換え蛋白質の発現―発現系の特徴と手順」(2008年)[16]や本稿の2.6項で後述する「#078 ディスポーザブルカルチャーバッグを利用したメタノール資化性酵母 Pichia pastoris による組換え蛋白質の分泌発現生産」(2014年)[17]などがある。
1.3 これまでに開発されてきたヒト Fas リガンド細胞外ドメイン(hFasLECD)の異種発現生産系
筆者は2012年に FasL や FasR を含むいわゆるデスリガンドならびにデスレセプターの細胞外ドメイン全般に関するそれまでの異種発現生産系の開発状況に関する総説を発表した[18]。ここでは、hFasLECD に限って、その後の進展を含めて概述する。hFasL 蛋白質は1回膜貫通 II 型の蛋白質であり、その構造遺伝子領域の解析から、N 末端側のドメイン(80残基)が細胞内に局在し、C 末端側の細胞外ドメイン(179残基)との間に22アミノ酸残基からなる膜貫通領域が存在することが明らかになっている(図1)。いわゆる分泌シグナル配列は N 末端部位には存在せず、翻訳された蛋白質が C 末端側ドメインを細胞外に表出した形で膜上に局在化するためのシグナルは中央の膜貫通部位に存在していると考えられる。完全な構造を持つ生物学的に活性な膜蛋白質としての hFasL は生理的条件下で3つの同一サブユニットからなるホモ3量体として存在するが、サブユニット当りジスルフィド結合1か所を含むその細胞外ドメイン部分のみを抜き出して生産した場合にも、正常な立体構造が形成された発現産物は3量体を形成する。細胞外ドメイン部分のみを活性型3量体として生産させるに当たっては、後述するようにそのN末端に分泌シグナル配列を人為的に付加し、酵母などの真核生物の分泌発現系を利用するのが有利である。具体的な発現生産系としては、これまでに P. pastoris[19,20]ならびに細胞性粘菌の一種であるキイロタマホコリカビ(Dictyostelium discoideum)を宿主とするもの[21]などが知られており、これらの宿主を使用した場合にはサブユニット当り3か所存在する N 結合型糖鎖付加部位への翻訳後修飾が生じると考えられる。
一方、分泌生産経路があまり高度には発達していない原核生物である大腸菌(Escherichia coli)を宿主とする菌体内発現系を用いる場合には糖鎖付加は起こらず、そのままでは正常な立体構造形成が生じ難いため、通常不溶性の発現産物として蓄積される[10,22]。先に述べた hDcR3 との複合体のX線結晶解析実験においては、大腸菌の菌体内発現系[23]により得られた不溶性産物を対象として試験管内で立体構造を再構成[24,25]させることにより調製された野生型ならびに変異型の可溶性 hFasLECD の試料が、昆虫由来の培養細胞の一種であるショウジョウバエの S2 細胞を用いて生産された hDcR3[26,27]との複合体形成に用いられた。当該論文中の記述によると野生型の hFasLECD 試料単独での結晶化は困難であり、真核生物で起こる糖鎖付加は単に水溶性を高めることに寄与しているだけではなく、凝集体の形成を抑止しているようである。
1.4 これまでに開発されてきた医療応用を目的とした hFasLECD 誘導体
細胞外ドメイン部分のみからなる可溶型の hFasLECD は単独の3量体分子の場合にはたとえ標的細胞表面上の FasR に結合しても、それだけでは効率良くアポトーシスを誘導することができない。また、ある種の低分子性抗がん物質によるアポトーシスの誘導においては、細胞表面を構成する脂質二重膜上でのラフト(筏)構造の形成等を介した FasR の会合が起こり、それに引き続くアポトーシスの実行を促進すると考えられている[28]。そのため、蛋白質工学を用いた分子デザインの観点から、細胞膜上で隣接する少なくとも2分子の FasR を会合させるべく、hFasLECD に改変を加え hFasLECD-hFasR 複合体のオリゴマー形成を促進させることを目的に各種の工夫が行われてきた。中でもヒトアディポネクチンのコラーゲン様ドメインが付加された誘導体である MegaFasL[29]は2分子の3量体型 hFasLECD からなる6量体構造を形成し、ヒトの悪性脳腫瘍の原因となるグリオーマ細胞やその幹細胞様細胞に対してアポトーシスを誘導する能力を示すことが明らかにされている[30,31]。MegaFasL はハムスター卵巣由来の CHO 細胞を用いた工業的生産が可能[32]であり、製品名 APO010(Oncology Venture 社)として各種のがん細胞に対する細胞死誘導作用が確認され、再発型あるいは難治型の多発性骨髄腫患者を対象とする第 I 相および第 II 相の臨床試験(Clinical Trial.gov, ID:NCT03196947)も行われた。また、インターロイキン6ファミリーに属するサイトカインの一種である白血病阻止因子レセプター gP190 の D1、Ig および D2 などのドメインの付加によるオリゴマー形成についても検討され[33]、Ig ドメインを付加したものは6量体ならびに12量体を形成し、ヒトの類表皮腫が移植された免疫不全マウスに対して延命効果を示すことが明らかになった。
一方、hFasLECD の治療用蛋白質としての開発を検討する上で、最も重要な点の一つは全身性投与の際に生じると考えられる肝臓を始めとする hFasR を介したアポトーシス誘導に対して感受性の高い正常な臓器への重篤な副作用の軽減である[34]。この課題を克服するために、標的細胞へのターゲティング能力を付与させるための数多くの工夫が施されてきた。代表的な戦略として、標的とするがん細胞表面上に豊富に発現する抗原に特異性を有する単鎖化可変部ドメイン(scFv)型の抗体フラグメントとの遺伝子レベルでの結合による融合蛋白質の調製が挙げられる。これまでにB 細胞を起源とするリンパ腫細胞上の CD20 抗原や T 細胞を起源とする白血病細胞上の CD7 抗原などをターゲットとして、HEK293 細胞や CHO 細胞などの哺乳動物由来の培養細胞で発現生産[35,36]された融合蛋白質が試験に供され、培養細胞ならびに実験動物個体において一定の増殖抑制効果が確認されている。この手法を用いて医療応用を目的として開発された hFasLECD 融合蛋白質についての主な例を表1にまとめて示す[37-50]。
2. hFasLECD 誘導体の Pichia pastoris 分泌発現生産系を用いた調製
2.1 分泌発現生産系の構築
筆者が本研究を開始する以前に、P. pastoris を用いた hFasLECD の分泌発現生産に関する先行研究として、大阪バイオサイエンス研究所と持田製薬株式会社の共同研究グループから、完全長の当該ドメインに対応する103番目から281番目までのアミノ酸残基を対象とした P. pastoris GS115 株を宿主とする研究結果が発表されていた[19]。1997年に J. Immunology 誌に発表されたこの研究で用いられた hFasLECD の分泌発現用遺伝子ユニットは、5’ 末端側から P. pastoris 由来のアルコールオキシダーゼ1(AOX-1)遺伝子のプロモーター配列、パン酵母(Saccharomyces cerevisiae)由来のα-接合因子遺伝子の分泌用プレプロシグナル配列、ヒト由来の cDNA を鋳型とし PCR 法により複製増幅された DNA フラグメントにより構成されている。またこの遺伝子ユニットを搭載するベクターとしてはヒスチジンに関する栄養要求性型選択マーカー遺伝子(HIS4)を含むプラスミドの一つである pPIC9 が用いられた。発現誘導の条件としては、P. pastoris を宿主としメタノールにより発現の誘導を行なう場合の代表的な培地である Buffered Methanol-complex Medium(通称:BMMY 培地)が使用され、30℃、48時間の培養条件下で OD600 = 100 という高濃度の培養開始時菌体密度条件下に実施された。この際の分泌生産量については、後に P. pastoris による蛋白質の発現生産に関する権威の一人である J.M. Cregg 博士らにより2000年の FEMS Microbiology Reviews 誌に発表された総説[51]の中で、100 mgL-1 という培養液の単位容積当りの生産量としてかなり高いレベルの値が記載されているが、これは先に述べた菌体密度の高さが一因となっているのかも知れない。以下に述べる研究はこの先行研究の結果を基盤として行ったものである。
筆者らの研究では、化学合成遺伝子を使用してヒト由来の溶菌酵素の一種であるヒトリゾチーム[52]ならびに小麦由来のレクチンの一種である小麦胚芽アグルチニン[53]を対象とした酵母を宿主とする異種蛋白質の分泌発現生産に関わってきた経緯から、hFasLECD の P. pastoris を用いた発現生産系の構築における構造遺伝子の調製においても同じく化学合成法を採用した[20]。この構造遺伝子の設計に当たっては酵母において多量に発現される同種蛋白質遺伝子において優先的に使用されているコドン[54]を主に使用した。一方、上記の先行研究の場合と同じく、宿主としては P. pastoris GS115 株を使用し、AOX-1 プロモーター配列とα-接合因子遺伝子のプレプロシグナル配列の後に野生型 hFasLECD の103番目から281番目までのアミノ酸残基をコードする遺伝子を連結させたユニットを構築した。構築した発現用遺伝子ユニットを抗生物質ゼオシン(Zeocin)に対する抵抗性遺伝子を選択用マーカーとして含む分泌発現用ベクターの一種である pPICZαA に搭載したものを用いて発現試験を行った[20]。発現の誘導は同じくBMMY 培地を使用し、29.5℃で実施した。残念ながら発現誘導開始時の菌体密度に関する詳細な記録が残っていないため正確に比較することは困難であるが、培養上清中の分泌生産レベルは 1〜2 mgL-1 程度と先行研究の場合と比べてはるかに低いものであった。
2.2 タグ配列付加ならびに糖鎖付加部位欠失の影響
発現生産量を上昇させるための最初の試みとしてタグ配列の付加を行った。発現産物の精製工程においてアフィニティークロマトグラフィーを利用する可能性を考慮して、FLAG(AspTyrLysAspAspAspAspLys)タグもしくは (His)6 タグを選択し、かつそれぞれに精製時の立体障害に起因する収量低下を回避するべく (Gly)5 からなるフレキシブルなスペーサー配列を介して構造遺伝子の N 末端側あるいは C 末端側に接続した計4種類のタグ付加型誘導体遺伝子を作成して分泌発現を試みた[20]。その結果、タグ配列を付加しない場合と比較して、N 末端側に FLAG-(Gly)5 配列を配置した場合にのみ分泌生産量の向上が認められた(図4)。この事例を含めて、以降本研究で検討した hFasLECD の各種誘導体における構造遺伝子ユニットの構成と培養上清の SDS-PAGE 分析により推定されたおよその分泌生産レベルの増減の関係を図4にまとめて示した。次に hFasLECD 遺伝子中に3箇所存在する N 結合型糖鎖付加部位を構成する Asn 残基(Asn184、Asn250 及び Asn260)に関する改変を試みた。 N 末端に FLAG-(Gly)5 タグ配列を付加した誘導体について N 末端側から順に1箇所、2箇所ならびに3箇所の Asn 残基をそれぞれ Gln 残基に変換して糖鎖付加部位を欠失させたものを作製し、野生型とともに SDS-PAGE 分析で目的とする発現産物のバンドの濃さにより比較したところ、野生型を含めた分泌生産量の比較では Asn184Gln 変異体が最も多く、精製試料の収量は約 5 mgL-1 となることが明らかとなった。
一方、同じくN末端側に付加した場合でも (His)6-(Gly)5 タグでは逆に分泌生産量は半減した。また、いずれのタグ配列でも C 末端側に付加した場合や3箇所の糖鎖結合部位の全ての Asn 残基に Gln 残基への変異を導入した場合には、目的とする発現産物の分泌生産は認められなくなり、これらの場合には分泌過程での hFasLECD を構成するペプチド鎖の正常な立体構造形成への重大な悪影響が生じていることが推測された。特に Asn260Gln 変異の導入効果は大きく、同じく1箇所の糖鎖結合部位でも Asn260 残基のみを残存させた場合には変異導入前の場合とほぼ同程度の分泌生産量が認められたのに対して、Asn250 残基のみを残存させた場合には5個の独立した形質転換体のシングルコロニーの培養上清を SDS-PAGE を用いて解析した限り、いずれについても目的物の生産量は極めて少ないか、あるいは一旦対応する発現産物の薄いバンドが出現したように思われるものでも培養を続ける間に消失し、分解や不溶化を起こしやすい少量の発現産物が分泌されたものと推測された(図4、未発表データ)。これらの結果は P. pastoris の分泌経路内での hFasLECD の正常な立体構造の形成や発現産物の安定性の維持に関して Asn260 への糖鎖の付加が極めて重要であることを現している。また、各部位に糖鎖が付加したものに対応する発現産物の SDS-PAGE 分析におけるそれぞれのバンドの濃さから判断して、少なくとも P. pastoris GS115 株を宿主とした場合には、糖鎖付加に関して Asn184 部位の欠失の影響は小さく、この部位に糖鎖が付加する割合は少ないこと、一方 Asn250 部位の欠失の影響は大きく、この部位への糖鎖付加はほぼ完全に生じることが明らかになった[20]。以上から、同じN結合型糖鎖付加部位でも立体構造上の位置の違いに応じて P. pastoris を宿主とした分泌発現生産に与える特徴や挙動が大きく異なることが示唆された。
hFasLECD の P. pastoris を宿主とした分泌生産において Asn260 に付加される糖鎖の構造的完全性が重要であることを探るために、BioGrammatics 社から市販され SuperMan5 株と名付けられている、酵母でのコア糖鎖部分から外側への糖鎖の伸長に関わる OCH1 遺伝子が欠損し Man5 と呼ばれるコア型糖鎖(Man5GlcNAc2)部分のみが付加される株[55]を宿主とする分泌発現を試みた。この SuperMan5 株を用いた系では、2.3項で後述する GS115 株を用いた系で最も分泌発現生産量が多かった変異体の一つである [Δ(103-138),Asn184Gln+Asn250Gln] 変異体について検討した場合でも、培養液を直接 SDS-PAGE で分析した限り明確な発現産物のバンドを検出することはできなかった(図4)(未発表データ)。この結果は、Asn260 位に付加するコア構造以外の部分を含めた糖鎖構造の完全性が P. pastoris の分泌経路における hFasLECD 分子の正常な立体構造形成の過程において極めて重要であり、その過程においてグルコシダーゼ I および II ならびにカルネキシン、カルレティキュリンなどのシャペロン機能を有するレクチン様分子が関与するコア型構造部分以外の糖鎖に対する認識[56]が本質的な役割を果たしていることを強く示唆している。不完全な構造の糖鎖を有するために正常な立体構造が形成されなかった hFasLECD 分子は、おそらく小胞体(ER)内での品質管理機構である小胞体関連分解(ER-associated degradation)による処理を受けリサイクル過程に回されたものと考えられる[57]。
当初、著者は hFasLECD 単体での立体構造解析のための結晶化を目指していたため、Asn184 残基のみ、あるいは Asn184 残基と Asn250 残基を同時に Gln に変換し N 結合型糖鎖付加部位をあらかじめ部分的に欠失させた変異体について、さらに化学構造の均一化を図るべく、Endo Hf グリコシダーゼ(NEB 社製)による残存する糖鎖のトリミングを行い、単一の N-アセチルグルコサミン残基への変換を試みた[58]。結果として、Asn250 残基に結合している糖鎖は短時間で容易に切断されるのに対し、Asn260 残基に結合した糖鎖の切断にはより多くの酵素量と反応時間を要することが明らかになった。この事実は先に述べた Asn260 残基に付加する糖鎖の hFasLECD の立体構造安定性への正の寄与と呼応するものである。また、[Δ(103-138),Asn184Gln+Asn250Gln] 変異体に残存する Asn260 残基に付加された糖鎖を Endo Hf で切断し N-アセチルグルコサミン1残基のみとしたものについて、結晶化条件探索のための実験に必要な濃度までの濃縮を試みたところ、一定の濃度に達した段階で沈殿物が生じ、不溶化を伴う凝集が起こりやすい生成物であった(未発表データ)。この現象は大腸菌菌体内で生産された糖鎖を持たない hFasLECD の発現産物を再構成して可溶化した試料が凝集を起こし易いという観察[10]とも一致している。残存する糖鎖を Endo Hf で切断した精製試料について MALDI-TOF 法による質量分析を用いた解析(アプロサイエンス社受託)を行ったところ、確かに予想された N-アセチルグルコサミン1残基のみが付加された均一な化学構造と一致した分析結果が得られたことから、P. pastoris GS115 株を宿主として分泌生産された発現産物は O 結合型糖鎖の付加などの他の翻訳後修飾は受けていないものと推察された[58]。
2.3 N 末端領域の除去が分泌生産量に与える効果
hFasLECD の構造遺伝子については、過去に哺乳動物細胞を用いて行われていた一連の欠失変異体の解析[59]から、N 末端部位のアミノ酸残基103番から138番目までの領域は FasR 結合活性の発現に必要な3量体構造を形成する上においては必須ではないことが既に明らかになっていた。この領域を除去して P. pastoris を宿主とした分泌生産を行った場合にどのような影響が生じるかを明らかにする目的で、上述した Asn184Gln 変異体ならびに [Asn184Gln+Asn250Gln] 二重変異体をベースとしてさらにこの領域に欠失変異 [Δ(103-138)] を追加した遺伝子を作成して発現生産を試みた[58]。その結果、興味深いことにいずれの変異体の場合にも大幅に分泌生産量が増加し(図4)、[Δ(103-138),Asn184Gln+Asn250Gln]変異体の場合、その精製収量はおよそ 24 mgL-1 に達することが明らかとなった。当該欠失領域の C 末端にはPro134Ser135Pro136Pro137Pro138 というプロリン残基が多数連続するアミノ酸配列が存在(図2)しており、その特異性から正常な立体構造を形成する際の律速段階となっている可能性がある。[Δ(103-138)] 変異体ではこの配列が除去されたために、P. pastoris の ER 内での効率的な立体構造形成が起こり大幅な分泌生産量の増加につながったのではないかと考えられる。
2.4 陽イオン交換クロマトグラフィーによる精製
2.1項で述べた P. pastoris を宿主とした先行研究においては、分泌生産された発現産物は高マンノース型糖鎖が付加された糖蛋白質であることを利用して、精製工程の第一段階に Con A アガロースによるアフィニティークロマトグラフィーが使用された[19]。一方、著者は研究開始当初から糖鎖のトリミングによる化学構造の均一化を計画していたため、原理的に糖鎖の有無に依存しない陽イオン交換クロマトグラフィーによる分泌発現産物の精製法を開発した。
野生型 hFasLECD の一次構造中には酸性アミノ酸残基である Asp 残基と Glu 残基を合わせた数に比べて塩基性アミノ酸残基である Lys 残基と Arg 残基を合わせた数の方が多く含まれ、アミノ酸残基103番から281番までの領域には、酸性残基が16個と塩基性残基が20個、アミノ酸残基139番から281番までの領域には、酸性残基が12個と塩基性残基が17個存在する(図2)。ExPASy Proteomics Server[60]を用いた計算によると等電点(pI)はそれぞれ理論上8.96ならびに9.15と予測された。予備実験の結果、幸いにして調製した各変異体は pH 5.5 付近でも安定に可溶性で存在することが明らかになったため、スルホプロピル基が坦持された陽イオン交換体である Hi-Trap S カラム、Resource S カラムあるいは Mono S カラム(いずれも GE ヘルスケア社製)を用いた分画精製を行った。具体的には培養上清を濃縮後、pH 5.3~5.6 の 50 mM 酢酸ナトリウム緩衝液への充分な置換を行った試料について、最初に Hi-Trap S カラムを用いたマニュアルのシリンジ操作での塩濃度を段階的に増加させた溶出による粗分画を行い、引き続きより分離能が高い Resource S カラムまたは Mono S カラムを用いた高速液体クロマトグラフィーシステムによる塩濃度を連続的に上昇させた溶出による分画を実施して、最終精製試料を調製した。
N 末端領域を欠失させ、かつ N 結合型糖鎖が付加される残基を Asn260 のみとした [Δ(103-138), Asn184Gln+Asn250Gln] 変異体は、精製工程におけるクロマトグラム上3箇所の N 結合型糖鎖付加部位を有する野生型の試料に比べてよりシャープな形状の目的物のピークが得られたことから、分泌生産量が多くより純度の高い試料の調製に有利であることとも併せて、以後の実験においてはこの変異体を各種誘導体作成のための基本的な変異体として使用することにした。また、陽イオン交換クロマトグラフィーを用いる精製に関連して、3.1項以降で詳述する部位特異的化学修飾体の調製を行った際に、一部のものについては、N 末端に付加した FLAG-GlyCys(Gly)4 タグ配列中に塩基性残基である Lys を新たに3残基挿入した FLAG-(Lys)3GlyCys(Gly)4 タグ配列を使用することにより、上記の Hi-Trap S カラムを用いた粗精製段階における分画前試料に関する緩衝液の置換をより簡略的に行えるようにすることで、精製工程における操作性を向上させることが可能であった[61]。
2.5 ヒト Fas レセプター細胞外ドメインに対する結合活性の確認
hFasL による標的細胞に対するアポトーシスの実行は、この蛋白質分子に対して現在知られている唯一のシグナル伝達実行型の受容体であるヒトFasレセプター細胞外ドメイン(hFasRECD)との特異的な結合を介して誘起される。したがって、P. pastoris を宿主として分泌生産された hFasLECD の誘導体ががん細胞などの標的細胞に対してアポトーシスによる細胞死誘導活性を示す潜在的能力を有するかどうかについては、基本的に hFasRECD に対する特異的な結合活性の有無により判断することが可能であり、本研究においてもこの結合活性評価試験用に適した hFasRECD を含む分子が必要であった。しかしながら、その目的に合う代表的な分子である hFasRECD とヒト IgG1 の Fc ドメインの融合蛋白質(hFasRECD-Fc)に関しては、研究用試薬としてマウス由来の培養細胞の一つである NS0 細胞を用いて発現生産された精製試料(R&D 社製)などが当時市販されていたものの、いずれも少量かつ高価であったため必ずしも容易に使用できる状況にはなかった。そのため、自ら試験用の試料を調製する必要に迫られ、先に2.1項で述べた hFasLECD 遺伝子を同じく酵母における頻用コドンを用いて化学合成により調製した hFasRECD 遺伝子と置き換えた形の発現用遺伝子ユニットを構築し、同じ手順により分泌発現を試みたが期待に反して培養液中に目的の発現産物の分泌を確認することはできなかった(未発表データ)。
そこで、先行研究として文献上の記載があったマウス由来の FasRECD とヒト IgG1 の Fc ドメインの融合蛋白質の例[62]にならい、昆虫細胞を使用した発現生産系[63]を用いて hFasRECD-Fc を調製する方策に転換した。その際には、有償の受託発現サービスを利用した。最初に、まず発現生産用に用いられる代表的な昆虫培養細胞の一つであるヨトウ蛾(Spodoptera frugipera)由来の sf9 細胞を用いる系[64](タカラバイオ社受託)で比較的高収量(6.7 mgL-1)の分泌発現が可能であることを確認した後、より生産量の増加が期待できるカイコ蛾(Bombyx mori)の幼虫の血リンパ液中への分泌生産[65](片倉工業社受託)を試みた。後者の系は良好に機能し、発現生産のためのバキュロウイルスを感染させた5齢幼虫由来の 26 ml の血リンパ液から 22.5 mg の精製試料が得られた。尚、カイコ蛾幼虫の血リンパ液中には内在性の夾雑蛋白質が多量に存在するため目的とする組換え体蛋白質の精製が困難になる場合があることが予想されるが、この点に関しても hFasRECD-Fc の場合、当該 Fc ドメインに対する高い特異性と充分な結合容量を兼ね備えたプロテイン G を坦持した市販のアフィニティー精製用カラム(Hi-Trap プロテイン G、GE ヘルスケア社製)が入手可能であったため、これに引き続く高分離能の陰イオン交換クロマトグラフィー用カラム(Resource Q、GE ヘルスケア社製)を用いた分画と組み合わせることにより簡便に精製試料の調製を行うことができた[66]。
hFasLECD の hFasRECD に対する特異的結合活性の評価試験での利用において hFasRECD-Fc の有利な点の一つは、市販のプロテイン A またはプロテイン G が坦持されたアガロースビーズや磁気ビーズを併せて用いることにより、共免疫沈降試験が実施可能であることである。この試験において、これまでに調製した hFasLECD の誘導体は後述する部位特異的化学修飾を施したものも含めていずれも hFasRECD-Fc と安定な複合体を形成して沈降物中に回収され、hFasRECD に対する強い結合活性を有することが確認されている。
また、hFasLECD と Fc ドメインが結合していない hFasRECD との複合体の形成について調べるため、hFasRECD-Fc における両ドメイン間のヒンジ領域にセリンプロテアーゼの一種であるトロンビン(Thrombin)による特異的認識切断サイトとして AlaAlaAlaProArgGlySerAla の配列を挿入した hFasRECD-T-Fc 型誘導体についてもカイコ幼虫を用いた分泌発現生産を行った[67]。この hFasRECD-T-Fc についても高レベルの発現生産が認められ、組換えカイコ幼虫由来の 25 ml の血リンパ液から 13.5 mg の精製試料が得られた。これをトロンビンにより切断後、切り離されたFcドメインをプロテイン G 坦持カラムにより吸着除去し、さらに素通り画分中に含まれる蛋白質を陽イオン交換カラム(Resource S)により分画することにより、hFasRECD 部分のみからなる精製試料を調製した。尚、hFasRECD 中にはトリプトファン残基が含まれないため、P. pastoris を用いて生産された hFasLECD 誘導体の単離された hFasRECD に対する溶液中での特異的結合については、両者を種々の割合で混合した試料についてのサイズ排除クロマトグラフィーを用いた分析を行い、215 nm と 280 nm の2種類の波長の吸光度を同時に検出することで確認した[67]。
さらに、hFasRECD-T-Fc 分子内の hFasRECD 中に存在する2箇所の N 結合型糖鎖付加部位に関してそれらを構成する Asn 残基を共に Gln 残基に変換することで欠失させた変異体についても、発現量の低下は認められたものの、同様にカイコ幼虫の血リンパ液中への分泌生産により調製することが可能であった[68]。また、N 末端タグ配列が付加されていない hFasLECD の [Asn184Gln+Asn250Gln] 二重変異体中に残存する糖鎖を Endo Hf を用いてトリミングした試料と上記の hFasRECD ドメイン領域内に糖鎖付加部位を持たない hFasRECD-Fc 変異体試料を用いて共免疫沈降試験を行った場合にも、当該リガンド–レセプター間の強い結合が確認された。この結果は、少なくとも試験管内での hFasLECD と hFasRECD の結合においてはそれぞれが有する糖鎖に由来する相互作用の寄与は少ないことを示唆している。
2.6 ディスポーザブルカルチャーバッグを用いた培養システムの開発
P. pastoris を宿主とする異種蛋白質の分泌生産実験においては、発現誘導用として P. pastoris 由来の AOX-1 遺伝子の強力なプロモーター遺伝子領域を、また分泌用シグナルとして S. cerevisiae 由来のα-接合因子遺伝子のプレプロシグナル配列を使用することが効率的な分泌発現生産用遺伝子ユニットを構成する上での代表的な戦略の一つになっている[11]。P. pastoris はメタノール資化性の酵母であり、メタノールを唯一の炭素源とした培養が可能である。その場合、代謝の第一段階であるメタノールのホルムアルデヒドへの変換のために多量のアルコールオキシダーゼが必要となるため、自ずから AOX-1 プロモーターの転写活性が高まる。また、その変換反応における基質の一つである酸素分子は、培地に対する溶解度が低くアルコールオキシダーゼに対する親和性は必ずしも高くない[69]ため、培養時の通気を良くすることによりその充分な供給を確保することが遺伝子発現の効率を高める上で極めて重要になる。
hFasLECD の発現生産系を構築するに当たって P. pastoris を宿主とする分泌発現系を選択した理由の一つに、小規模な研究室において使用可能なレベルの装置や培養規模でも立体構造解析のための結晶化探索実験や部位特異的な化学修飾を手段とする高機能化を目指した実験に必要な量の精製試料を調製できる可能性があることが挙げられる。本研究では、市販の微生物培養用の全容積 5 L あるいは 10 L のポリプロピレン製のディスポーザブルカルチャーバッグ(藤森工業社製)を培養容器として用い、ダイアフラム式の小型ポンプにより強制的な通気を実施する培養システムを新たに開発して使用した(図5a、5b)[68]。このシステムにおいて、実際に使用可能な培地量は各カルチャーバッグ全容積の半分以下であるため、上記のバッグを使用した場合、それぞれ一回で 2.5 L あるいは 5 L 規模までの培養が実施可能である。それまでに使用してきた 3 L 容量のガラス製バッフル付きフラスコ(培養液量、500 mL/本)を用いた培養システムとカルチャーバッグを使用するシステムでの分泌生産量を、N 末端にタグ配列が付加されていない hFasLECD の [Δ(103-138),Asn184Gln+Asn250Gln] 変異体を対象として最終的な精製物の収量で比較したところ、単位培養容積当り約3倍の増加が認められた[68]。この培養システムは、これまでフラスコ培養の際に使用してきた恒温回転振とう式の培養機をそのまま利用可能で、発現誘導用の培地についても同じ組成のものが使用できるためガラス製バッフル付きフラスコを用いた系から簡便に移行することができる。また、本研究で使用したディスポーザブルカルチャーバッグは比較的安価(5 L 容量および 10 L 容量共に2万円弱/5セット)で入手可能であった[17]。次項で述べる部位特異的化学修飾体の調製実験では反応に供するためにできるだけ多量の N 末端タグ配列中にシステイン残基を導入した誘導体の精製試料が必要であったため、その取得を目的とした発現生産実験においては専らこのシステムを使用した。陽イオン交換クロマトグラフィーを用いた精製工程に供するための培養上清中の目的とする発現産物の効率的な濃縮ならびに脱塩操作においては、並列に連結した複数個のペリスタ式ポンプのそれぞれにポリエーテルスルホン製の限外濾過膜(排除限界分子量: 10 kDa)を内蔵したタンジェンシャルフロー(TFF)型の小型カセット(ポール社製)を接続した装置の利用が有効であった(図5c)。
3. hFasLECD の部位特異的化学修飾体の調製とキャラクタリゼーション
3.1 N 末端タグ配列中にシステイン残基を有する誘導体の設計と発現生産
hFasLECD の部位特異的化学修飾を手段とする高機能化の戦略を考えるに当たっては、化学修飾の導入後もこの蛋白質の医療応用上最も有用な生物活性の一つである hFasRECD に対する特異的な結合に基づくアポトーシス誘導活性を保持させることを念頭に置く必要がある。そのためには、hFasRECD との結合の際の相互作用様式を事前に立体構造の観点から検討しておくことが重要である。1.1項で述べたように、一次構造の相同性から hFasLECD-hFasRECD 複合体と立体構造が類似していると推定されるデスリガンド‐デスレセプターの組み合わせであるヒト TNFβ-ヒト55kd TNF レセプター細胞外ドメイン複合体の X 線結晶構造解析結果[70]などをもとに、hFasLECD(サブユニット3量体)は hFasRECD(単量体)と1:3のモル比で複合体を形成することが比較的古くから推定されていた。この推定の正しさは、hFasLECD とデコイ型レセプターである hDcR3 の複合体の立体構造が発表されるに至り[10]、ほぼ確定的になったものと考えられる。
この複合体の立体構造解析結果によると hFasLECD の N 末端部位は hDcR3 との結合の際に互いが直接接触する界面部位から三次元的に離れた位置に存在している(図3)。そのため、N 末端に親水性の高いタグ配列を付加した場合、タグ配列を構成するポリペプチド鎖は hFasRECD に対する結合を阻害することなく溶媒中に露出して存在することがあらかじめ予測できた。この予測に基づいて、2.2項で記した hFasLECD 本体(アミノ酸残基番号139-281)の N 末端に付加した FLAG-(Gly)5 タグ中の連続した Gly 残基の領域内に部位特異的化学修飾反応の足がかりとなるシステイン(Cys)残基を挿入することにより FLAG-GlyCys(Gly)4 とした。その際に Cys 残基を最初の Gly 残基のあとに配置した理由は、当該 Cys 残基に対して化学修飾により付加される分子が FLAG タグ配列固有の機能になるべく影響を及ぼすことなく、かつ挿入した Cys 残基が hFasLECD 本体からできるだけ離れた位置にくるようにすることで溶媒中に露出しやすくなり化学修飾に関する反応性が高まることを期待したためである。
この N 末端タグ配列内に Cys 残基を導入した誘導体について、当該 Cys 残基を含まない誘導体の場合と同一の方法を用いて P. pastoris GS115 株を宿主とした発現を試みたところ、3量体中の2つのサブユニットの一部がそれぞれのタグ配列内に存在する Cys 残基の間でジスルフィド(SS)結合を形成して培養液中に分泌されることが非還元条件下での SDS-PAGE 分析により明らかになった[68]。しかし、この部分的にサブユニット間の SS 結合が形成された発現産物についてもそれまでのタグ配列内に Cys 残基を含まない誘導体の場合と同様な方法で精製をすることが可能であった。
野生型の hFasLECD 分子中には立体構造上 Cys202-Cys233 間にサブユニット当り1か所の SS 結合が元来存在する(図3)。そのため、このドメイン内部に埋もれた SS 結合を保持した状態でN末端部位に連結したタグ配列内に存在する Cys 残基に対して部位特異的にかつ効率的に化学修飾を施すためには、タグ配列中の Cys 残基の側鎖のみを反応性の SH 基を持つように活性化した状態に保つ必要がある。この目的を達成するためにはトリス(2-カルボキシエチル)ホスフィン(TCEP)の使用が有効であった。TCEP はその比較的大きな分子サイズのため、2-メルカプトエタノール(2ME)やジチオスレイトール(DTT)などのチオール性還元剤を使用した場合に比べて、適切な濃度や反応温度を設定することで、蛋白質分子内部に埋もれた SS 結合に影響を与えることなく、より選択的に溶媒中に露出した SS 結合のみを還元して反応性を有する SH 基とすることができる[71]。また、反応機構上も 2ME や DTT を用いた場合のような SS 結合の交換反応が起こらないため、基本的には過剰の TCEP を除去することなくマレイミド化合物等による修飾反応に供することが可能である。P. pastoris を宿主として分泌生産された上記の N 末端タグ配列内に Cys 残基を含む誘導体の場合、室温(25℃)、pH 5.5 の条件下で、10~20 mM の TCEP を1時間程度作用させることで選択的な SH 基の活性化の目的を達成することができた。他方、過剰の TCEP がマレイミド基を持つ化学修飾用試薬と反応し、ペプチド中に含まれる Cys 残基とマレイミド基を有する色素分子との反応を阻害する可能性を指摘する報告もある[72]。そのため、次の段階の修飾反応に移る前に、マレイミド化合物との反応に適した pH 6.4 の 25 mM のリン酸緩衝液への交換を兼ねて、念のため事前に PD-10 カラム(GE ヘルスケア社製)を用いた分画により余分な TCEP をできるだけ除いた直後にマレイミド化合物との反応に供した。その際に 2~5 mM 程度の EDTA を共存させることにより SS 結合の再生を防いだ。尚、過剰の TCEP を除いた後の試料を非還元条件の SDS-PAGE 分析に供した際に、SS 結合が再生された産物に対応すると考えられる分子量の位置に薄いバンドが認められたが、上記のマレイミド化合物との反応後にはこのバンドは確認できなかった[68]ことから、このバンドは SDS-PAGE 分析の操作過程において pH の上昇などが原因で SS 結合が一部再生することにより検出されたものと推察された。
3.2 部位特異的化学修飾体の調製法の検討
これまでに hFasLECD に関する部位特異的化学修飾体(部位特異的化学修飾による他分子との共有結合体)の調製法として、上記のタグ配列中に Cys 残基が含まれる誘導体を対象として、i) 大過剰の目的とする修飾用分子のマレイミド基含有誘導体をチオール–エン反応により直接付加させる方法[73]、ならびに ii) 最初に6-メチルテトラジン(MTZ)基またはトランスシクロオクテン(TCO)基を含むマレイミド化合物とのチオール–エン付加反応により末端に TCO 基あるいは MTZ 基を持つスペーサー用分子を結合させた hFasLECD 誘導体を調製し、一旦単離した後、引き続き目的とする修飾用分子との間で TCO 基–MTZ 基間の逆電子要請ディールスアルダー型(iEDDA)付加反応により連結を行う方法[61]の2種類について検討した(図6)。図7に本研究において各蛋白質分子に対して末端に TCO 基ならびに MTZ 基を含むエチレングリコールオリゴマー型スペーサーを導入する際ならびに付加反応後残存する未反応の TCO 基および MTZ 基のクエンチング反応を行う際に使用した市販試薬(いずれも Click Chemistry Tools 社製)の化学構造式を示した。また、引用文献61の Fig.1 にこれらの試薬を用いて調製した TCO 基あるいは MTZ 基を末端に有する各タンパク質分子に関する誘導体の構造式が記載されているので併せてご参照いただきたい。
化学修飾法による分子の付加は必ずしも定量的に達成できるとは限らない。チオール–エン反応による直接付加の事例として、N 末端に FLAG-GlyCys(Gly)4 タグ配列を有する hFasLECD の [Δ(103-138),Asn184Gln+Asn250Gln] 変異体に対して大過剰のフルオレセイン-5-マレイミドを用いて反応を行なった際の修飾率は精製試料中に導入された蛍光色素団固有の吸光度値から83%と推算された[73]。一方、iEDDA 付加反応を用いた事例として、N 末端に FLAG-(Lys)3GlyCys(Gly)4 のタグ配列を持つ hFasLECD の [Δ(103-138),Asn184Gln+Asn250Gln] 変異体に対して、20倍過剰モル当量の3単位のエチレングリコールユニットの両端に TCO 基とマレイミド基を有する化合物(TCO-PEG3-MAL)(図7)を反応させて調製した hFasLECD の TCO 基含有誘導体(hFasLECD-PEG3-TCO)と MTZ 基ならびにメトキシ基を末端に持つ分子量約 5 kDa のポリエチレングリコール(PEG)の誘導体(mPEG-MTZ, 5 kDa)(図7)との反応を実施した。この付加反応においては1.0~1.5倍過剰モル当量程度の mPEG-MTZ, 5 kDaの使用量で付加体の収率は飽和に達してほぼ一定になることが反応混合物の SDS-PAGE 分析により確認されたが、この場合にも修飾率は80%程度であった[61]。
通常マレイミド基を含む修飾用分子を直接用いる反応の場合は被修飾蛋白質分子に対して過剰なモル比の修飾用試薬が必要になるが、1段階反応での付加が行なえるため全体として目的とする分子による修飾率が2段階反応を行う場合に比べてより高くなる。そのため、修飾用分子のマレイミド誘導体が比較的容易に入手可能な場合に適している。一方、第2段階として使用される TCO 基–MTZ 基間の付加反応は Cys 残基–マレイミド基間の反応を用いる場合に比べて、被修飾分子に対してより少ないモル比の修飾用分子を用いても室温下で円滑に反応が進行する。そのため、より貴重な低分子化合物や機能性蛋白質分子など一般的には潤沢な量が得難い分子を被修飾用分子としたい場合に適していると考えられる。具体的には、高い修飾率を目指して反応を行ないたい場合、Cys 残基とマレイミド基の反応においては典型的には25倍過剰モル当量程度の修飾用試薬を使用する必要がある[71]のに対し、TCO 基と MTZ 基の付加反応の場合には 1~数 mgmL-1 程度の濃度の蛋白質分子の溶液について1.0~1.5倍過剰モル当量程度の被修飾分子を添加した場合でも高い修飾率の達成が期待できる。
また、反応前の試料を保存する際に、Cys 残基やマレイミド基の場合は通常多くの蛋白質分子が安定に存在できる pH 7~8 付近の生理的緩衝液条件下において、それぞれ空気酸化による SS 結合の形成ならびに加水分解によるマレアミド酸基への変換が起こりやすく、これらに由来する官能基の不活性化の可能性を常に考慮する必要がある。これに対し、TCO 基や MTZ 基の場合には同様な条件下での安定性が高く、通常は試料溶液を-20℃以下で凍結保存すれば長期間にわたって大きな活性の低下を危惧せずに使用可能である。そのため、モノクローナル抗体などを始めとする貴重な蛋白質を修飾用分子として用いる場合には特に有用であると考えられる。
3.3 低分子性蛍光色素による化学修飾体の調製
これまでに上述した N 末端タグ配列中に Cys 残基を導入した2種類の hFasLECD 誘導体に関する低分子性蛍光色素による化学修飾体として、フルオレセイン-5-マレイミドを用いた1段階反応法による修飾体の調製[73]、ならびに TCO 基–MTZ 基間の付加反応を用いた2段階反応法によるスルホ-Cy3 基を導入した修飾体の調製[61]を行った。調製した全ての蛍光性化学修飾体は hFasRECD-Fc(または hFasRECD-T-Fc)とプロテイン A 担持アガロース(ロシュ社製)(またはプロテイン G 坦持磁気ビーズ、バイオラッド社製)を使用した共免疫沈降試験により、非修飾体と同等な hFasRECD 結合活性を有することが明らかとなった。加えて、各蛍光色素団に由来する可視領域の吸収極大(フルオレセイン基:495 nm、スルホ-Cy3 基:550 nm)付近の吸光度を指標とするサイズ排除クロマトグラフィーを用いた複合体形成確認試験においても明確な hFasRECD 結合活性が確認できた。これらの結果は、先述した hFasLECD–hDcR3 間複合体の立体構造解析情報に基づく部位特異的化学修飾の戦略が hFasRECD 結合活性の保持において有効であったことを示している。
調製した蛍光性化学修飾体はいずれもそれぞれの蛍光色素団に特徴的な励起ならびに蛍光スペクトルを示したが、精製物試料の紫外・可視領域の吸収スペクトルから推算された hFasLECD サブユニット3量体当りの各蛍光色素基の平均導入数は、1段階反応により導入したフルオレセイン基の場合2.5であったのに対し、2段階反応で導入したスルホ-Cy3基の場合には、スルホ-Cy3 基を含む分子側の官能基として MTZ 基ならびに TCO 基を用いて導入した各修飾体でそれぞれ1.5および1.6であり、あらかじめ予想されたように付加反応の段階数の増加による蛍光色素基の導入率の低下が認められた。また、サイズ排除クロマトグラフィーを用いた各蛍光性化学修飾体と hFasRECD-Fc との複合体形成試験において、TCO 基含有誘導体の形でスルホ-Cy3 基を導入した修飾体では、同様な分析条件にもかかわらず、上記のフルオレセイン基修飾体や MTZ 基含有誘導体の形でスルホ-Cy3 基を導入した蛍光性修飾体を用いた場合に比べて、hFasLECD の蛍光色素修飾体と hFasRECD-Fc の複合体がカラム内でわずかながら解離しやすい傾向が認められた[61,73]。この結果は TCO 基含有誘導体の形でスルホ-Cy3 基を導入した場合には、修飾により hFasLECD と hFasRECD-Fc の相互作用における会合強度が立体障害等による何らかの理由により低下した可能性を示唆するものである。そのため、次項で述べる他の機能性蛋白質分子を修飾用分子として用いた架橋体の作成実験においては、修飾用蛋白質分子の MTZ 基含有誘導体と hFasLECD-PEG3-TCO の間の iEDDA 付加反応を使用した。
3.4 他の機能性蛋白質分子との架橋体の調製
筆者はこれまでに hFasLECD-PEG3-TCO(約 60 kDa)に関する他の機能性蛋白質による修飾のモデル実験として、ニワトリ卵白由来のアビジン(Avi、約 66 kDa)ならびにウサギ IgG 由来の Fab’ ドメイン(rFab’、約 55 kDa)を使用した架橋体の調製について検討した[61]。図8aに hFasLECD とこれらの蛋白質のおよその分子の大きさと形の比較のために PDB に登録されているデータをもとに描画した立体構造(但し、rFab’ については Fab 部分のみ)を示した。
4つの等価なサブユニットからなる Avi 分子中には1サブユニットあたり Lys 残基が9個存在する。Avi に関する MTZ 基含有誘導体(Avi-PEG4-MTZ)は、サブユニット当り8倍過剰モル当量の4単位のエチレングリコールオリゴマーユニットの両端に MTZ 基ならびにスルホ-N-ヒドロキシスクシンイミドエステル基を有する化合物(MTZ-PEG4-sulfo NHS ester)(図7)を 0.1 M の炭酸水素ナトリウム水溶液(pH 8.3)中、室温(24℃)で反応させることにより調製した。そのため、原則的には分子表面に存在する不特定な Lys 残基の位置に MTZ 基が導入されると考えられる。尚、Lumiprobe 社の技術資料 “NHS ester labeling of amino biomolecules”[74]によると、このような反応条件を用いた場合一般的な蛋白質やペプチドでは平均1個の Lys 残基が修飾されるが、後に hFasLECD-PEG3-TCO との反応により得られた架橋体の SDS-PAGE 分析による解析結果は1サブユニットに複数の MTZ 基が結合しているものが少量含まれていることを示唆していた[61]。一方、rFab’ に関する MTZ 基含有誘導体(rFab’-PEG4-MTZ)については、rFab’ と MTZ-PEG4-MAL との反応により調製した。最初に、ウサギの血清からプロテイン A が担持されたアフィニティーカラムを用いて精製された全 IgG 分子をブタ胃由来のペプシンにより切断して F(ab’)2 ドメインとしたものをさらに37℃で 10 mM の EDTA を含む 10 mM の2-メルカプトエチルアミンを用いて処理[75]することにより、C 末端近傍に単一の反応性 Cys 残基を有する rFab’ を得た。引き続き、この rFab’ ドメインを直ちに大過剰の4単位のエチレングリコールオリゴマーユニットの両端に MTZ 基ならびにマレイミド基を有する化合物(MTZ-PEG4-MAL)(図7)と反応させることにより rFab’-PEG4-MTZ を調製した。したがって、MTZ 基の導入位置は C 末端部位に限られ、導入数は1個と考えられる。
hFasLECD-PEG3-TCO と Avi-PEG4-MTZ または rFab’-PEG4-MTZ とのいくつかの混合モル比条件下での反応物について、150 mM の NaCl を含む 50 mM Tris-HCl(pH7.5)を溶出用緩衝液とし、Superdex Increase 200 10/300カラム(GE ヘルスケア社製)を用いて行ったサイズ排除クロマトグラフィー分析に関する代表的なプロフィールを図9に示した。hFasLECD-PEG3-TCO(2.5 mgmL-1)とそれぞれ1.0、1.2、1.5及び3.0倍過剰モル当量の Avi-PEG4-MTZ(4.3 mgmL-1)を混合し、28℃で1時間静置した反応物を分析した結果、いずれの混合比の場合にも非架橋体試料の溶出位置に比べて高分子量側の領域に明確に分離したいくつかのピークが出現した。新たなピークが出現したクロマトグラム領域に関しては、1:1架橋体と考えられる最大のピークを含め、各ピーク間の相対的な高さの比や分離パターンなどのプロフィールには上記の4種類の反応条件間で本質的に大きな差は認められなかった[61](図9a)。また、上記のクロマトグラムにおいては、hFasLECD-Avi の1:1架橋体の分子量は理論上約130 kDaであるにもかかわらず分子量サイズマーカーの一つとして使用した球状蛋白質であるアルドラーゼ(158 kDa)のピーク溶出位置よりも若干高分子量側の位置にピークが認められた。これは、1:1架橋体の形状が単純な球状ではなく類似した分子量の2つの球状蛋白質分子がエチレングリコールオリゴマー鎖を含むスペーサーにより連結された亜鈴様の形状(図8b)であることに由来するものと推量される。架橋反応後 Avi 分子上に残存する未反応の MTZ 基を大過剰の TCO 基を含有する1級アミン化合物(TCO-NH2・HCl)(図7)を用いてクエンチングした試料について上記の1:1架橋体に対応するピーク画分を分取した。この分画物試料は hFasRECD-Fc に対する強い結合活性を保持すると同時に、ビオチンを坦持したヤギ由来抗ウサギIgG (H&L) 抗体(アブカム社製)を用いた共免疫沈降試験ならびにビオチン誘導体化された蛍光色素分子である ATTO495-ビオチン(ATTO-TEC 社製)やビオチン-PEG5K-フルオレセイン(Nanocs 社製)との混合物について hFasLECD-Avi に結合していない過剰な蛍光色素分子をサイズ排除クロマトグラフィー分画で除去した精製複合体試料の分光学的解析を行うことにより、強いビオチン結合活性を有することが確認できた。ビオチン誘導体化蛍光色素分子との複合体の分光学的性質に関して、前者の場合、蛍光色素団と Avi 分子の間の距離が短いため両者間の相互作用に由来すると考えられる蛍光色素団の吸収スペクトルの極大波長の変化を伴う著しい消光現象が観察された[61]。一方、後者では蛍光色素団と Avi 分子の間に約 5 kDa のポリエチレングリコール鎖によるスペーサーが存在しているためこのような現象は起こらず、元来の蛍光強度を有する複合体が形成された(未発表データ)。
他方、hFasLECD-PEG3-TCO(2.5 mgmL-1)とそれぞれ1.0、2.0、3.0及び5.0倍過剰モル当量の rFab’-PEG4-MTZ(2.3 mgmL-1)を混合し、25℃で1時間静置した反応混合物を分析した結果、Avi-PEG4-MTZ の場合と同様に非架橋体の溶出位置に比べて高分子量側の領域に新たに分離したピークが出現した。Avi-PEG4-MTZ の場合と異なる点は、rFab’-PEG4-MTZ の使用モル数を増加するにつれて最初は1:1架橋体と考えられる成分が主ピークであったものが、徐々に1:2架橋体ならびに1:3架橋体と考えられるより高分子量側のピークに置き換わっていったことである[61]。すなわち、等モル当量の rFab’-PEG4-MTZ を使用した場合には1:1架橋体のピークが主であったものが、3倍過剰モル当量を使用した場合には1:1架橋体と1:2架橋体のピークの高さがほぼ同じになり、さらに5倍過剰モル当量に増加させた場合には1:1架橋体のピークはさらに減少し1:2架橋体ならびに1:3架橋体のピークが主成分となった(図9b)。これらのうち、等モル当量ならびに5倍過剰モル当量での架橋反応を行った後、hFasLECD-PEG3-TCO 分子上に残存する未反応の TCO 基を大過剰の MTZ 基を含む一級アミン化合物(MTZ-PEG4-NH2・HCl)(図7)を用いてクエンチング反応処理を行ない、各反応混合物から、1:1架橋体単独の画分ならびに1:2架橋体と1:3架橋体が主に含まれる混合物の画分をそれぞれ分取した。hFasRECD-Fc とプロテイン G 担持磁気ビーズを用いて行なった共免疫沈降試験の結果、いずれの分画物試料についても hFasRECD に対する強い結合活性を保持していることが確認できた。また、ウサギ IgG 全分子を坦持したアガロース担体を用いてアフィニティー精製されたヤギ由来抗ウサギ IgG (H&L) ポリクローナル抗体(アブカム社製)を使用した共免疫沈降試験により、この抗体により特異的に認識されるウサギ IgG 由来の Fab’ 断片が確かに結合していることを確認した[61]。
サイズ排除クロマトグラフィーによる分析結果から、Avi-PEG4-MTZ あるいは rFab’-PEG4-MTZ のいずれを使用した場合にも hFasLECD-PEG3-TCO との室温(25~28℃)下1時間処理後の等モル反応混合物には未だかなり多量(それぞれ45%および62%)の非架橋体が残存し、後者については反応時間を2倍に延長した場合にもその割合はほとんど変化しないことを確認した。この結果はたとえ効率的なクリック化学反応を利用したとしても、高分子量の蛋白質分子間の架橋反応の場合には立体障害等の観点から反応収率には一定の上限が存在する可能性を示している。この点に関しては、両蛋白質分子間の連結に使用するスペーサー長をより長くするなどの工夫により架橋体の収量を増加させることが可能になるかも知れない。
3.5 共有結合構造の同定と細胞死誘導活性の評価
これまでに3.4項で述べた hFasLECD-PEG3-TCO(サブユニット3量体)と Avi-PEG4-MTZ(サブユニット4量体)の間の付加反応により得られた1:1架橋体(図8b)の分画物試料を用いて、それぞれの蛋白質のサブユニットが解離する変性条件下での MALDI-TOF 質量分析法による構成コンポーネントの同定を目的とした確認試験(アプロサイエンス社受託)を行なった。シナピン酸とウシ由来血清アルブミン(約66 kDa)の+1価から+5価のピークの質量電荷比(m/z)値をそれぞれイオン化用マトリックスならびに m/z ピーク値に関する外部標準とした解析の結果、各蛋白質分子中の結合に関与していないサブユニット由来のシグナル(ピーク m/z 値:Avi-PEG4-MTZ サブユニット、15895.5; hFasLECD-PEG3-TCOサブユニット、20698.9)に加えて、両蛋白質分子のサブユニットが TCO 基–MTZ 基間の付加反応により共有結合したコンポーネントに由来する構造に対応するシグナル(ピーク m/z 値:36854.0)を同定することができた[2]。
一方、細胞死誘導活性については、ヒト結腸がん由来の株化細胞の1種である HT-29 細胞を被検細胞として、架橋用の抗 FLAG 配列 M2 モノクローナル抗体(Sigma 社製)の存在および非存在条件下における FLAG-GlyCys(Gly)4-hFasLECD の N-エチルマレイミド修飾体に関する評価、ならびにヒトインターフェロンガンマ(IFN-γ)による前処理の存在および非存在条件下での FLAG-GlyCys(Gly)4-hFasLECD に関するフルオレセイン5-マレイミド修飾体および上記の hFasLECD-PEG3-TCO と Avi-PEG4-MTZ の1:1架橋体の2種類の試料に関する評価を MTT アッセイ法による細胞生存率測定試験(生体分子計測研究所社受託)により行なった[2]。いずれの場合にも hFasLECD に関する部位特異的化学修飾体試料のみの場合には有意な細胞死誘導活性は検出されなかった。この結果は生体内において膜タンパク質である hFasL からマトリックスメタロプロテアーゼの一種である MMP7 による切断により生成することが知られている可溶型の hFasLECD が非常に弱いアポトーシス誘導活性しか示さないという実験結果[76]と一致している。他方、上記のようなアポトーシス誘導を増強させるための処理を施した場合、前者では 2 μgmL-1 の架橋用抗体共存下で100または 1000 ngmL-1 の hFasLECD 試料を用いた72時間の処理により約50%への細胞生存率の低下が[68]、また後者については、100 IUmL-1 の IFN-γ による24時間の前処理存在条件下において、いずれの試料を使用した場合にも100または 1000 ngmL-1 の hFasLECD 化学修飾体による72時間の処理によりほぼ全ての被検細胞の死滅が観測された(図10)[2]。
上記の抗 FLAG 配列抗体を用いる架橋処理あるいは IFN-γ を用いる前処理を施した条件下における hFasLECD の各部位特異的化学修飾体による細胞死誘導後の被検細胞を光学顕微鏡により観察したところ、いずれの場合についてもアポトーシス誘導の結果生じたと考えられる擬円形に収縮した形態の細胞の特徴を示していた(図10)[1,2]。前者の場合、抗体による架橋により少なくとも2組以上の hFasLECD と hFasR の複合体が近接して形成される[29]。一方、後者に関しては、IFN-γ はヒト体内において hFasL と同様にナチュラルキラー(NK)細胞ならびに細胞障害性 T リンパ球(CTL)細胞から産生されるサイトカインとして標的細胞上の特異的受容体を介した生体防御活性の発現に関わっていることが知られており[77]、標的細胞表面上の hFasR 分子の増加や局在化を誘起することにより hFasL によるアポトーシス誘導に対する感受性を高めることにも寄与していると考えられている[28,78,79]。本研究で得られた実験結果は、可溶型の hFasLECD を用いた場合でも標的細胞表面に発現する FasR との複合体をある一定以上の密度で形成させることにより効率的なアポトーシスによる細胞死を誘導することが可能である、というこれまでの研究に基づく作業仮説[80]を改めて支持するものである。加えて、後者の系においては hFasL の主な供給源である NK 細胞や CTL 細胞から分泌生産される IFN-γ を利用していることから、前者のような元来ヒト体内には存在しない FLAG タグ配列を認識する架橋用抗体分子を用いる場合に比べて、より生体内で生じているアポトーシスに近い機序での細胞死の誘導がなされているのではないかと推定される。
4. 今後の展望
上述したように、hFasLECD の P. pastoris を宿主とする各種誘導体の発現生産ならびに発現産物に関する部位特異的化学修飾体の調製を手段とする高機能化を目指した分子デザインは、未だようやく基本的な方法論が構築されつつある段階である。2010年に日本インターフェロン・サイトカイン学会より出版された書籍「サイトカインハンティング」にも詳しく記述されているように、日常ヒトの免疫系を巧みに制御している各種の重要なサイトカイン分子の発見や同定には、日本人研究者が深く関わったものが多い[4]。とりわけ本稿で対象とした hFasL は、1.1項でも述べたように標的細胞に対するアポトーシスの誘導という医療応用上有用な特異な機能を有している[5]。そのため、発見当初から単にそのヒト体内での生理学的ならびに病理学的役割の面からの興味に留まらず、がんや自己免疫疾患など薬物による治療という観点からは未だ医療上の目標が充分に達成されたとは言い難い重大な疾病を対象とした臨床応用が強く意識されると共に、期待も高かったのではないかと思われる[81,82]。
しかしながら、初期に行われた実験用マウスを用いた全身性投与実験の際に生じた劇症肝炎の発症が主因と考えられる死亡事例[19,83]からも推察されるように、機能性蛋白質素子として hFasLECD 単体やアゴニスト型抗 hFasR モノクローナル抗体のみを用いた治療法の開発はかなり困難を伴うのではないかと考えられる。この課題を解決するための糸口の一つとして、これまでにもがん細胞等への特異的なターゲティングのための scFv 型抗体フラグメントとの遺伝子レベルでの結合を手段とした融合蛋白質の調製を始めとする hFasLECD に関する各種の誘導体の設計、開発が試みられてきた(表1)。本稿で紹介した部位特異的化学修飾法による融合蛋白質分子の調製は、原理的には scFv 型フラグメントだけでなく全長分子を含む他の各種の形態の抗体分子等にも適用可能ではないかと推測され、新規な形態の機能性素子成分を含む治療用融合蛋白質分子の創製につながると考えられる。また、hFasLECD はマウス由来の FasR に対しても特異的に結合し、アポトーシス誘導活性を示すことが知られている[84]。P. pastoris で生産された可溶型 hFasLECD は、マウス FasR を発現するリンパ球由来の W4 細胞に対する細胞障害活性に関する試験管内での試験においては当該レセプターに対するアゴニスト型モノクローナル抗体の一つである Jo2 に比べて少なくとも20倍以上の高い比活性を有することが示されており、またマウスを用いた肝臓への毒性に関する生体内での試験ではその障害活性の程度は Jo2 に比べて50分の1以下であることも明らかになっている[19,34]。マウスを用いたこれらの実験結果はヒトを対象とした細胞死誘導型治療用蛋白質の開発を目指す上でのアゴニスト型抗 hFasR モノクローナル抗体に対する hFasLECD の優位性を暗に示すものであるが、適切な部位特異的化学修飾の適用により正常組織への重篤な副作用がさらに軽減されることが望まれる。
アビジンとの架橋体は各種のビオチンを坦持した分子と強く結合すると考えられ、多様な機能性分子や材料と安定な複合体を形成させることも可能である。また、細胞表面に表出する hFasR 分子は IFN-γ 以外にもウイルスの感染[82]、臨床的に用いられている抗がん剤[85]、γ線照射[86]あるいは高濃度のグルコース[87]等の作用により有意に増加することが知られている。この現象は、3.5項で例示したように、単独では弱い可溶性 hFasLECD 誘導体の標的細胞に対する細胞死誘導活性を増強させる手段として実際の疾病治療法の開発においても応用できる可能性を秘めているように思われる。血清などの各種体液中に含まれる可溶型の hFasRECD は将来的ながんによる死亡[88]、糖尿病や心血管病の発症増加[89]あるいはある種の急性腎臓病の進展予後[90]などに関するリスク因子として、各種の疾病に関わる予測のためのバイオマーカーとしての有用性が指摘されている。また、hFasL に結合するもう一つの特異的レセプターであり、体内でのがんの増殖や炎症反応に関する有用なバイオマーカーとしても知られている hDcR3[91]についても臨床上の固形がん患者の予後の推定に利用できることが示唆されている[92]。hFasRECD や hDcR3 のみを選択的に認識できる hFasLECD の部位特異的化学修飾体は疾病の診断用システムや治療薬の開発のための評価用システムを構築する際の分子ツールとしての利用も可能ではないだろうか。
P. pastoris を宿主とする発現生産系は、これまでに多種類の異種組換え蛋白質に関する大量調製の実績が豊富にあり高い確率で哺乳動物由来の培養細胞を用いる発現系に比べてより短期間かつ低コストでの目的蛋白質の生産が可能という、産業応用上の利点を有している[13,15]。また、哺乳動物細胞を用いる発現生産の場合のようなヒトに感染する可能性のある病原性ウイルスの混入についても危険性は少ないものと思われる。他方、付加される糖鎖構造が高マンノース型であり哺乳動物細胞由来のものとは異なることから、この糖鎖構造の違いに起因する免疫原性の惹起などの副作用が心配されるが、近年では哺乳動物細胞由来のものと類似した複合型糖鎖の付加が可能な株の開発も進んできた[93]。さらに現在では、知的財産面での制約についても、ATUM 社から野生型株に加えて PEP4 や PRB1 などの液胞プロテアーゼに関する欠損株を含む発現生産に適した数種類の “IP-Free Pichia strains” やそれらを用いる蛋白質の生産に適した発現ベクターが市販されており(コスモバイオ社扱い)、これらの製品を利用して製造した蛋白質の商用目的での使用についてはライセンスの取得を必要としないようである[94]。
近年、蛋白質分子の部位特異的化学修飾に適した生体直交型(bioorthogonal)の化学反応が種々開発されてきている[95]。中でも、本研究において使用した MTZ 基を含むジエン分子と TCO 基を含むジエノフィル分子との間での iEDDA 付加反応は、非常に高い反応速度定数(10~102 M-1s-1)を有し、かつ各官能基が通常蛋白質分子が変性を起こしにくい生理的 pH の緩衝液条件下において適度な化学的安定性を兼ね備えているため、とりわけ蛋白質分子間の架橋反応として大変利用価値が高いと考えられる[96,97]。一方、高分子量の蛋白質分子間の部位特異的化学架橋体を調製する場合、目的とする架橋体を高収量で経済的に得るためには反応条件に関する予備的検討が重要と思われる。最終的に精密な糖鎖構造を考慮に入れた哺乳動物細胞発現系で生産した被化学修飾用蛋白質が求められる場合においても、その前段階としての条件探索的研究では試験に供するための精製試料は多量に必要になると予想される。この点でも P. pastoris を宿主とする発現生産実験は比較的安価に実施可能であるため有用であろう。本稿で紹介した hFasLECD 誘導体の P. pastoris を宿主とした発現生産とその部位特異的化学修飾体の調製が、近い将来に実際の医療面での課題解決を図る上での有効な手段に発展することを期待したい。
5. 謝辞
蛋白質分子の立体構造図は日本蛋白質構造データバンク(PDBj)に登録されている座標データならびに PDBj から提供されているグラフィクス描画プログラム jV を用いて、また化学構造式は ACD 社のソフトウェア ChemSketch(Freeware)2016.1.1 を使用して作成しました。ここに記して謝意を表します。最後に、本総説中に記載した研究の遂行に当り多大な御協力を賜りました研究支援関連企業の担当者の方々、本稿の執筆を御奨励いただくと共に発表に際して大変お世話になりました産業技術総合研究所・バイオメディカル研究部門長、近江谷克裕博士ならびに同部門蛋白質デザイン研究グループ長、広田潔憲博士を始めとする関係者の皆様に深く感謝致します。
6. 引用文献
- Muraki, M., AIMS Bioengineering, 5, 39–62 (2018)
- Muraki, M. & Hirota, K., BMC Res. Notes, 11, 395 (2018)
- FASLG Fas ligand [Homo sapiens (human)]
- 長田 重一、米原 伸, サイトカインハンティング, 第8章 G-CSF, Fas, 253–67, 日本インターフェロン・サイトカイン学会編, 京都大学学術出版会 (2010)
- 須田 貴司, シリーズ・バイオサイエンスの新世紀 第6巻 細胞の誕生と死, 第3章 細胞死, ② FasとFasリガンド, 143–59, 長田 重一、山本 雅/日本生化学会編, 共立出版 (2001)
- Wajant, H. ed., Medical Intelligence Unit, Fas Signaling, Landes Bioscience, Georgetown / Springer Science+Business Media, New York (2006)
- Yi, F. et al., Trend. Mol. Med., 24, 642–53 (2018)
- Bodmer, LC. et al., Trends Biochem. Sci., 27, 19–26 (2002)
- Pitti, RM. et al., Nature, 396, 699–703 (1998)
- Liu, W. et al., Structure, 24, 2016–23 (2016)
- Ahmad, M. et al., Appl. Microbiol. Biotechnol., 98, 5301–17 (2014)
- Prabhu, AA. et al., Curr. Pharma. Biotechnol., 18, 1200–23 (2017)
- Pichia protocols (ed. Higgins DR. & Cregg JM.), Methods in Molecular Biology, 103 (Ser. Ed. Walker, JM), Humana Press, Totowa (1998)
- Pichia protocols Second Edition (ed. Cregg JM.), Methods in Molecular Biology, 389 (Ser. Ed. Walker, JM), Humana Press, Totowa (2007)
- 内藤 忠相、杉山 賢司、目的別で選べるタンパク質発現プロトコール、第3章 3.酵母、③タンパク質の発現、162–71, 永田 恭介、奥脇 暢 編、羊土社 (2010)
- 櫻井 一正、蛋白質科学会アーカイブ、1, e018 (2008)
- 村木 三智郎、蛋白質科学会アーカイブ、7, e078 (2014)
- Muraki, M., Prot. Pept. Lett., 19, 867–79 (2012)
- Tanaka, M. et al., J. Immunol., 158, 2303–09 (1997)
- Muraki, M., Prot. Expr. Purif., 50, 137–46 (2006)
- Luo, Z. et al., Biochem. Eng. J., 62, 86–91 (2012)
- Sun, KH. et al., Enzyme Microb. Technol., 36, 527–34 (2005)
- 萩原 義久、蛋白質科学会アーカイブ、1, e001 (2008)
- 浜田 寛之、松岡 常吉、蛋白質科学会アーカイブ, 1, e035 (2008)
- 梅津 光央、蛋白質科学会アーカイブ, 1, e038 (2008)
- Liu, W. et al., Mol. Biotechnol., 57, 914–22 (2015)
- 柴田 俊生 他、蛋白質科学会アーカイブ, 2, e052 (2009)
- Mollinedo, F. & Gajate, C., Drug Resist. Updates, 9, 51–73 (2006)
- Holler, N. et al., Mol. Cell. Biol., 23, 1428–40 (2003)
- Eisele, G. et al., Neuro Oncol., 13, 155–64 (2011)
- Eisele, G. et al., Anticancer Res., 33, 3563–71 (2013)
- http://www.oncologyventure.com/pipeline/apo010/
- Daburon, S. et al., Plos One, 8, e54000 (2013)
- Timmer, T. et al., J. Pathol., 196, 125–34 (2002)
- 橋口 隆生 他、蛋白質科学会アーカイブ、1, e017 (2008)
- 浅野 竜太郎, 蛋白質科学会アーカイブ, 2, e050 (2009)
- Samel, D. et al., J. Biol. Chem., 278, 32077–82 (2003)
- Bremer, E. et al., Cancer Res., 68, 597–604 (2008)
- Bremer, E. et al., Blood, 107, 2863–70 (2006)
- Chien, MH. et al., Mol. Cancer Ther., 16, 1102–13 (2017)
- Chan, DV. et al., J. Biomed. Sci., 20, 16 (2013)
- Hemmerle, T. et al., J. Biotechnol., 172, 73–6 (2014)
- Orbach, A. et al., Am. J. Pathol., 177, 3159–68 (2010)
- Aronin, A. et al., J. Hematol. Oncol., 7, 64 (2014)
- Zhang, W. et al., Mol. Immunol., 50, 150–9 (2012)
- Shi, W. et al., Graefe’s Arch. Clin. Exp. Ophthalmol., 245, 1691–7 (2007)
- Feng, YG. et al., Transplant. Proc., 37, 2379–81 (2005)
- Kitano, H. et al., J. Gene. Med., 14, 642–50 (2012)
- Morello, A. et al., Plos One, 8, e73375 (2013)
- Franke, DDH. et al., Mol. Immunol., 44, 2884–92 (2007)
- Cereghino, JL. & Cregg, JM., FEMS Microbiol. Rev., 24, 45–66 (2000)
- Jigami, Y. et al., Gene, 43, 273–9 (1986)
- Nagahora, H. et al., Eur. J. Biochem., 210, 989–97 (1992)
- Ikemura, T., Mol. Biol. Evol., 2, 13–34 (1985)
- Jacobs, PP. et al., Nat. Protoc., 4, 58–70 (2009)
- Helenius, A. & Aebi, M., Annu. Rev. Biochem., 73, 1019–49 (2004)
- Pfeffer, M. et al., Appl. Microbiol. Biotechnol., 93, 2503–12 (2012)
- Muraki, M., Prot. Expr. Purif., 60, 205–13 (2008)
- Orlinick, JR. et al., J. Biol. Chem., 272, 32221–9 (1997)
- Gasteiger, E. et al., Protein Identification and Analysis Tools on the ExPAsy Server (ed Walker JM.) 571–607, Humana Press (2005)
- Muraki, M. & Hirota, K., BMC Biotechnol., 17, 56 (2017)
- Mahiou, J. et al., Biochem. J., 330, 1051–8 (1998)
- 久武 幸司、福田 綾、目的別で選べるタンパク質発現プロトコール、第3章 2.昆虫細胞、120–44, 永田 恭介、奥脇 暢 編、羊土社 (2010)
- 前川 憲一、蛋白質科学会アーカイブ, 1, e022 (2008)
- 本庄 栄二郎、 蛋白質科学会アーカイブ, 1, e029 (2008)
- Muraki, M. & Honda, S., Prot. Expr. Purif., 73, 209–16 (2010)
- Muraki, M & Honda, S., Prot. Expr. Purif., 80, 102–9 (2011)
- Muraki, M., BMC Biotechnol., 14:19 (2014)
- Couderc, R. & Baratti, J., Agric. Biol. Chem., 44, 2279–89 (1980)
- Banner, DW. Et al., Cell, 73, 431–45 (1993)
- Hermanson, GT., Bioconjugate Techniques (Third Edition), Academic Press (2013)
- Shafer, DE. Et al., Anal. Biochem., 282, 161–4 (2000)
- Muraki, M., SpringerPlus, 5:997 (2016)
- https://www.lumiprobe.com/protocols/nhs-ester-labeling.pdf
- 石川 榮治, 生物化学実験法 27 酵素標識法, 第Ⅲ章, 31–50, 瓜谷 郁三 他編、学会出版センター (1991)
- Schneider, P. et al., J. Exp. Med., 187, 1205–13 (1998)
- Schroder, K. et al., J. Leukoc. Biol., 75, 163–89 (2004)
- Xu, X. et al., Cancer Res., 58, 2832–37 (1998)
- Schwartzberg, LS. et al., Clin. Cancer Res., 8, 2488–98 (2002)
- Thorburn, A., Cell. Signal., 16, 139–44 (2004)
- Nagata, S., Cell, 88, 355–65 (1997)
- 中西 義信, 蛋白質 核酸 酵素, 41, 2552–8 (1996)
- Ogasawara, J. et al., Nature, 364, 806–9 (1993)
- Takahashi, T. et al., Int. Immunol., 6, 1567–74 (1994)
- Micheau, O. et al., J. Natl. Cancer Inst., 89, 783–9 (1997)
- Reap, EA. et al., Proc. Natl. Acad. Sci. USA, 94, 5750–5 (1997)
- Maedler, K. et al., Diabetes, 50, 1683–90 (2001)
- Tamakoshi, A. et al., Int. J. Cancer, 123, 1913–6 (2008)
- Mattisson, IY. et al., EBioMedicine, 26, 187–97 (2017)
- Bhatraju, PK. et al., Crit. Care, 21, 217 (2017)
- Hsieh, SL. & Lin, WW., J. Biomed. Sci., 24, 39 (2017)
- Ge, H. et al., Clin. Chim. Acta, 481, 126–31 (2018)
- Liu, L. et al., Biologicals, 39, 205–10 (2011)
- 1.5. License for Pichia Strains, ATUM 社 TERMS & CONDITIONS
- Oliveira, BL., Chem. Soc. Rev., 46, 4895–950 (2017)
- Wu, H. & Devaraj, NK., Top. Curr. Chem., 374, 3 (2016)
- Mayer, S. & Lang, K., Synthesis-Stuttgart, 49, 830–48 (2017)
