

概要
その生物学的重要性にも関わらず、膜タンパク質は精製時の発現量や安定性が可溶性タンパク質よりも低く、精製試料の調製が可溶性タンパク質よりも困難であることが多い。そういった問題を克服するべく、膜タンパク質の構造解析における有力な手法として、GFPタグを用いたFluorescence-Detection Size-Exclusion Chromatography(GFP-FSEC)法が幅広く用いられている。GFP-FSEC法では、対象タンパク質をGFP融合タンパク質として発現させ、可溶化試料に対して蛍光検出機を備えたHPLCを用いてゲルろ過クロマトグラフィーを行う。そして、得られたHPLCのチャートに基づき、構造解析に適した発現系評価が行われる。GFP蛍光は高い感度と特異性を持つため、数mL程度の少量培養からの試料を未精製にて発現系の評価を行うことが可能となる。つまり、GFP-FSEC法を用いることで安価かつより短いタイムスケールでの発現系の良しあしの評価を行うことが可能となる。本稿では、膜タンパク質の発現系評価を目的としたGFP-FSEC法の実際について述べる。また、この2024年改訂版では、2010年の初版後に発表された近年の発展的手法についても関連文献を原稿の末尾に紹介する。
イントロダクション
近年の数多くの技術的進歩により、構造決定されたタンパク質の数は指数関数的に増加し、現在ではProtein Data Bank(PDB)に登録されている構造数は20万を超えるに至った(2024年1月現在)。従来構造解析が困難とされた膜タンパク質についても7000を超えるなど大幅な増加をみせている。その背景にはクライオ電顕をはじめとする構造解析手法のブレークスルーのみならず、以下に述べるような膜タンパク質の発現・精製における各種問題を克服するような試料調製段階での技術進歩も大きい。
組み換えタンパク質としての膜タンパク質の構造解析における大きな問題点として
- 組み換えタンパク質としての発現量の乏しさ
- 界面活性剤による可溶化後の膜タンパク質の安定性の低さ
の2点が挙げられる。しかしながら、このような膜タンパク質の一般的な傾向とは逆に、
- 組み換えタンパク質としての比較的良好な発現量
- 界面活性剤溶液中での安定性
という上記2点の特徴を持った発現系が構造解析を行う上では望ましい。実際、構造解析に成功した膜タンパク質の多くはこれらの特性を満たしている。2013年前後からこの10年で広く普及したクライオ電顕による構造解析の場合もこの傾向は大なり小なり共通している。
そのような構造解析に望ましい発現系の同定のためには、構造解析対象とするタンパク質の複数のホモログについて発現トライアルを行い、構造解析に適したホモログを同定するというストラテジーが従来広く用いられてきた。例えば、筆者らのグループが行ったマグネシウム輸送体MgtEの構造解析では7種類のMgtEホモログをクローニングし、それらについて発現系の評価を行い、最終的に高度好熱菌Thermus thermophilus由来MgtEを「構造解析に適したホモログ」として同定している(1)。しかしながら、膜タンパク質の発現量、安定性、均一性の評価には、多くの場合、500 mLないし1 L程度の中規模の培養スケールにて発現・精製を行う必要がある。そのため、多数のホモログに対してこれを行うためには、かなりの時間とコストが要求され、膜タンパク質の構造解析の律速段階となっている。また、2024年現在では幅広く行われるようになった哺乳類由来の膜タンパク質のクライオ電顕による構造解析の場合も、哺乳類の生物種、界面活性剤条件の探索などを初期段階の実験をスモールスケールで迅速に行えることは望ましい。
そのような問題に対する評価手法の1つとして、GFPタグを利用したFluorescence-Detection Size-Exclusion Chromatography(GFP-FSEC)法がある(2)。GFP-FSEC法では、対象タンパク質をGFP融合タンパク質として発現させ、可溶化試料に対して蛍光検出機を備えたHPLCを用いてゲルろ過クロマトグラフィーを行う。対象となるタンパク質の発現量については、ゲルろ過クロマトグラフィーにおけるピークのGFP蛍光強度の高さにより評価される。また、試料の安定性については、ゲルろ過クロマトグラフィーにおけるピークの形状により評価される。すなわち、「構造解析に理想的な発現系」はゲルろ過クロマトグラフィーにおいて鋭く、高い単一ピークを示す。GFP蛍光は高い感度と特異性を持つため、数mL程度の少量培養からの試料を未精製にて発現系の評価を行うことが可能である。そのため、GFP-FSEC法は安価かつより短いタイムスケールでの発現系評価を行うことを可能とし、膜タンパク質の構造解析の律速段階のブレイクスルーとして過去20年近く幅広く用いられてきた。実際、本稿で筆者らが参考としたGFP-FSEC法の原著論文(2)を発表したGouaux博士のグループはGFP-FSEC法を利用し、膜輸送体の構造解析において大きな成功をおさめている(3–7)。その後、広く普及したクライオ電顕による構造解析でもGFP-FSEC法は広く利用されている。本稿では、膜タンパク質の発現系評価を目的としたGFP-FSEC法の実際と2010年の初稿発表後の進展について述べる。こうした質の高い膜のタンパク質の試料調製は、構造解析のみならず、各種生化学・生物物理の実験においても有用なものである。
装置・器具・試薬
HPLC(製品更新に注意)
- システムコントローラCBM-20A(島津製作所)
- 送液ユニットLC-20AD(島津製作所)
- オートサンプラSIL-20AC(島津製作所)
- カラムオーブンCTO-20A(島津製作所)
* システムを低温室に設置する場合は不要 - UV-VIS検出器SPD-20A(島津製作所)
- 蛍光検出器RF-20Axs(島津製作所)
- オンラインデガッサDGU-20A5(島津製作所)
- LC-20AD用低圧グラディエントユニット(島津製作所)
* 本稿で述べるGFP-FSEC法には必須ではない。 - 高圧流路切換バルブFCV-12AH(島津製作所)
- ゲルろ過クロマトグラフィー用カラム(Superose 6 increase 10/300 GLもしくは5/150 GL、Superdex 200 10/300 increase GLもしくは5.150 GL(Cytiva))
- 各種バッファー(破砕バッファー、可溶化バッファー、ゲルろ過クロマトグラフィーバッファー)
原核生物の膜タンパク質の発現プラスミドおよび発現ホスト
- GFP融合タンパク質発現プラスミド
* Gouaux博士よりpNGFP-BC(N末端GFP融合タンパク質発現用)およびpCGFP-BC(C末端GFP融合タンパク質発現用)の譲渡を受け、それらに対して目的遺伝子をクローニングした。ベクターマップについては筆者らが参考とした原著論文(2)を参照されたい。 - 大腸菌C41(DE3)コンピテントセル(Overexpress)
* 他のBL21系の大腸菌についても使用可能である
また、大腸菌を用いた組み換えタンパク質の発現については蛋白質科学会アーカイブ「T7プロモータを利用した大腸菌による組み換え蛋白質の発現」など他稿にその詳細な記述があり、培養および発現のための装置・器具・試薬や実験の詳細についてはそちらを参照されたい。
超遠心(製品更新に注意)
- Optima MAX-TL ultracentrifuge(Beckman)
- TLA-100.3 rotor(Beckman)
実験手順
- 第1日
- 目的プラスミドのC41(DE3)への形質転換
- 第2日
- 前培養
- 第3日
- 本培養と発現誘導
- 第4日
- 膜画分の調製
- 可溶化
- HPLC分析
* 膜画分については-80度保管が可能であるため、5.および6.については後日行うことも可能である
実験の詳細
第1日
1. 目的プラスミドによるC41(DE3)の形質転換
ヒートショック法もしくはエレクトロポレーションにて、目的とする遺伝子を含むプラスミドによりC41(DE3)コンピテントセルを形質転換する。アンピシリンを含むLBプレート上で37℃一晩静置。多数の試料について形質転換を行う場合は、ヒートショック法が簡便である。
第2日
2. 前培養
前日の各LBプレートから、コロニーを取り、3 mLスケールのLB培地に植菌して37℃で一晩振盪培養を行う。GFP-FSEC法では多数の培養試料を同時に取り扱うことが多く、多数の試料についてIPTGによる発現誘導の際のタイミングを合わせるためには、各培養試料間の濁度をそろえる必要がある。そのため前培養を行う。
第3日
3. 本培養と発現誘導
前日の前培養試料から培地を0.1 mL取り、3 mLスケールのLB培地に植菌し、37℃で振盪培養を開始する。数時間ほどで培養液の600 nm付近のODが0.5程となるのでIPTGを加え、発現誘導を行う。試料数が数十にも及ぶ場合、全ての試料のODを測定するのは現実的ではないため、代表していくつかのODを測定するとよい。加えるIPTG濃度やIPTG添加後の培養時間、温度については条件検討が必要である。初期条件としては、目的タンパク質一種あたり0.5 mM IPTG濃度にて
- 37℃、3時間培養
- 37℃、一晩培養(16–20時間程度)
- 20℃、一晩培養(16–20時間程度)
の3条件を試みることを推奨する。筆者の経験では20℃培養が効果的であることが多い。これは膜タンパク質の過剰発現が細胞に対して毒性を持つことに起因する可能性が指摘されており、低温培養が有効な事例がいくつか報告されている(8)。
第4日
4. 膜画分の調製
以降の作業は全て氷上もしくは4℃下にて行う。前日の3 mLスケールの培養試料について、2回に分けて1.5 mLエッペンドルフチューブに集菌する。各チューブに0.5 mLの破砕用バッファーを加え、超音波破砕機を用いて、超音波破砕を行う。破砕バッファーの組成についての1例としては、150 mM NaCl、50 mM HEPES pH 7.0、1 mM PMSF、1 mM DTTが挙げられる。破砕後、16,000 × gにて10分遠心し、上清を新しい超遠心用のチューブに移し、TLA100.3にて40,000 rpm(87,000 × g)1時間超遠心を行う。超遠心後、エッペンドルフチューブの底に微量の膜画分があることを確認する。多数の試料を取り扱い、膜画分の調製に時間がかかる場合は、調製後、試料を−80℃に保管し、以降の作業を後日行ってもよい。
5. 可溶化
膜画分を含むエッペンドルフチューブに0.2 mLの可溶化バッファー(150 mM NaCl,50 mM HEPES pH 7.0,2%ドデシルマルトシド(DDM),1 mM DTT)を加える。他に膜タンパク質の構造解析でしばしば用いられる界面活性剤としては、従来用いられてきたデシルマルトシドやオクチルグルコシドに加え、LMNGやGDN、Digitoninなどもクライオ電顕の普及に伴い広く用いられている。バッファーを加えた際、膜画分はエッペンの底に付着しているため、市販の1.5 mLチューブ用のホモジェナイザーを用いて膜画分を懸濁させる。その後、ローテーターを用いて1時間ほど4℃で振盪させ、可溶化を行う。可溶化後、TLA100.3にて再度40,000 rpm(87,000 × g)、1時間超遠心を行い、上清を新しい1.5 mLチューブに移す。
6. HPLC分析
HPLC分析についても4℃下で行う。ゲルろ過クロマトグラフィー用バッファー(150 mM NaCl,50 mM HEPES pH 7.0,0.05% DDM)により平衡化されたゲルろ過クロマトグラフィーカラムに対して、オートサンプラーを用いて可溶化試料を0.1 mL注入する。この際、流速はメーカー設定に従い、1回の注入あたり1.2カラムボリュームほど溶出させる。蛍光検出機RF-20AXs(島津製作所)の標準設定は、参考とした原著論文(2)のGFPuvの場合、励起/蛍光波長 = 395/507 μmとなっているが、用いる蛍光タンパク質の種類に応じて適宜調整が必要である。各種設定は、参考とした原著論文(2)のグループらの設定に基づく。HPLCの溶出プロファイルについては、ASCII形式で出力することが可能であるため、EXCELを用いてグラフを作成することができる。図1に、筆者らのグループによって得られたHPLCチャートの1例を示す。
工夫とコツ
発現ターゲットの探索
複数のホモログをクローニングし、発現トライアルを行う場合、原核生物由来の輸送体の場合、有望なターゲットみつけるためには、最低でも10種類ほどを試したほうがよい。原核生物のゲノムもしくは菌株については、理化学研究所の微生物材料開発室から比較的安価に入手可能である。しかしながら、近年顕著に増えてきた真核生物の膜タンパク質の構造解析、特に哺乳類の膜タンパク質の構造解析の場合、配列相同性の多様性を確保しつつ様々な生物種に由来するホモログを確保することは困難である。その一方、近年の膜タンパク質の構造解析の主な手法となったクライオ電顕では以前ほどタンパク質の安定性は重視されない。そのため、多数のホモログスクリーニングではなく、界面活性剤条件の探索で十分なことが多い。そのため、比較的少数の哺乳類ホモログに対し、ドデシルマルトシドだけではなく、LMNG、GDN、Digitoninなどの安定化作用の強い界面活性剤を用いて条件探索することを推奨する。また、そういった際の安定性評価の定量的な手法として、FSEC-TS法(9)が発表されており、詳細は当該論文を参照されたい。
蛍光タンパク質タグの位置と種類
同じ遺伝子であっても、GFPタグの箇所(N末、C末)とで、得られるHPLCチャートに有意な差があることがあるため(2)、可能な限り、N末、C末それぞれのGFP融合発現系を作成することを推奨する。また、N末端、C末端ともに細胞外側に位置している場合、蛍光タンパク質を直接融合させないタイプの新しい手法、FSEC-Nb法も近年報告されている(10)。この手法では、ALFAペプチドタグとの融合タンパク質として標的を発現させ、ALFAタグに対するナノ抗体との融合タンパク質として発現させたGFPで標的を標識させることで、標的の膜タンパク質の発現評価を行っている。また、GFP以外の赤色や青色蛍光タンパク質を用いることで、膜タンパク質の複合体形成をFSEC法によって評価する試みも報告されている(11)。
オートサンプラーの注意点
オートサンプラーを用いることにより、一度に多数の試料について分析を行うことが可能であるが、その場合、最初に注入された試料と最後に注入された試料とでは、可溶化からHPLC分析までの時間に大きな差ができることになる。そのため、後半に注入された試料については、時間経過による分解や凝集などが問題となる可能性がある。よって、カラムボリュームが24 mlの標準的なカラムを用いる場合の各試料について整合性のある結果を得るためには、1回の可溶化で分析する試料数は多くても1晩分(15から20程度)に留め、さらなる分析については後日、別に可溶化を行ったほうがよい。もしくはカラムボリュームが3 ml程度のいわゆるミニカラムを用いる場合はより多くのサンプル処理が一晩で可能である。
GFPを利用した発現量およびピーク形状の評価
HPLC分析の結果、得られる値は相対的な蛍光強度であり、それ自体では絶対的な発現量の見積もりを行うことはできない。そのため、GFP単体の精製試料もしくは発現量が知られているGFP融合膜タンパク質の発現系を用いて、発現量の指標とするのがよい。また、ピーク形状に関しては、結晶構造解析が主な構造決定手法であった際は単一の鋭いピーク形状であることが非常に重視されたが、クライオ電顕による構造解析の際はピーク形状はそこまで重視されていない。しかしながら、排除体積(void)の限界を超えるような凝集が大量にみられる試料はやはり構造解析には適しておらず、そうではない範囲の比較的良好なピーク形状がみられる試料をFSEC法によって選択した上で、実際にクライオ電顕上でサンプル評価をすることが重要となってきている。
文献
- Hattori, M., et al., Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 63, 682–684 (2007)
- Kawate, T. & Gouaux, E. Structure, 14, 673–681 (2006)
- Yamashita, A., et al., Nature, 437, 215–223 (2005)
- Jasti, J., et al., Nature, 449, 316–323 (2007)
- Kawate, T., et al., Nature, 460, 592–598 (2009)
- Shaffer, P. L., et al., Science, 325, 1010–1014 (2009)
- Sobolevsky, A. I., et al., Nature, 462, 745–756 (2009)
- Wang, D. N. et al., Biochim. Biophys. Acta., 1610, 23–36 (2003)
- Hattori, M., et al., Structure., 20, 1293–1299 (2012)
- Jin, F., et al., Commun Biol., 4, 366 (2021)
- Parcej, D., et al., PLoS One., 25, e67112 (2013)
改訂履歴
2024年1月19日 改訂
- 著者所属の情報の更新。
- 近年のFSEC関連技術の進展を紹介したことにあわせてタイトルの更新。
- 近年のクライオ電顕技術の発達を踏まえ、概要およびイントロダクションを更新。
- 装置・器具・試薬中の型番情報等を更新。
- 「実験の詳細」中の推奨する界面活性剤条件等を更新。
- 近年のクライオ電顕技術やFSEC技術の発達等を踏まえ「工夫とコツ」を更新。
- 引用文献の増加に対して文献情報を更新。
-
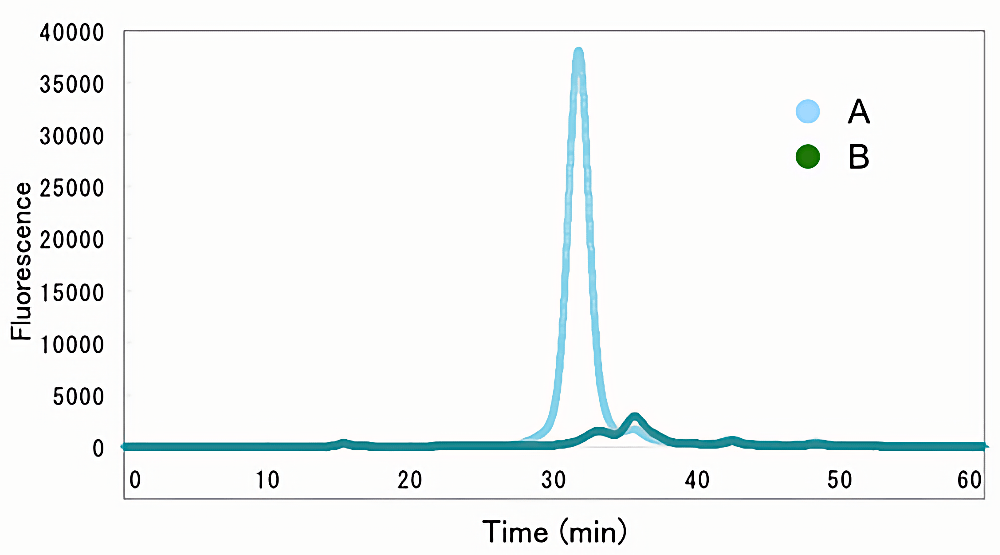
図1:GFP-FSEC法におけるHPLCチャートの例
縦軸および横軸はそれぞれ蛍光強度と時間(分)に対応する。発現系AとBを比較すると、Aは、強い蛍光強度と単一の鋭いピークを示しており、有望な発現系であると考えられる。
