

概要
Transdirect は既存の無細胞タンパク質合成試薬キットとは異なり、バキュロウイルスを用いた組み換えタンパク質生産にも広く利用されている Sf21 昆虫細胞(Spodoptera frugiperda 卵巣細胞由来)の抽出液を利用している。抽出方法や抽出時に添加する試薬を工夫することで、抽出液は無色透明で複数回の凍結融解に対しても安定となっている。また本キットには、高効率にタンパク質合成を行うための専用発現ベクター pTD1(Accession Number:AB194742)を添付している。このベクターには Sf21 細胞抽出液に適した、バキュロウイルスの一種である MnNPV(Malacosma neustria nucleopolyhedrovirus)のポリヘドリン遺伝子5’非翻訳領域中の配列が翻訳促進配列として導入されているほか、mRNA 合成用 T7 プロモーター配列、マルチプルクローニングサイト(MCS)、polyA 領域などの mRNA 合成からタンパク質合成に関わる全ての因子が含まれている。さらに、反応バッファーは複数のモデルタンパク質を用いて、特に K+ や Mg++ イオン濃度を調整することで基本的にどのタンパク質でも最適な合成が行えるように至適化されている(1,2)。
このように Transdirect は、安定な細胞抽出液と至適化された発現ベクター、反応バッファーとの組合せによって、非常に効率のよいタンパク質合成を行うことができる(反応液1 mL あたり30-50 μg)世界で初めての昆虫細胞由来の試薬キットとなっている。この合成量は、これまで唯一の動物細胞由来であったウサギ網状赤血球の系と比較して10倍以上の値である。
本プロトコルでは、この Transdirect を用いた目的タンパク質取得までの標準的な工程について述べる。このプロトコルに従い、これまでに数多くのタンパク質を取得し、質量分析装置を用いた翻訳後修飾の解析などに成功している(3,4)。
装置・器具・試薬
- サーマルサイクラー
- 恒温槽(振とう培養が可能なもの)
- 分光光度計
- 遠心分離装置
-
蛍光イメージアナライザー(レーザーベース)
- NICKTM Columns(GE社、17-0855-01)
- PD-10(GE社、17-0851-01)
-
遠心濃縮フィルター
- 滅菌蒸留水
- フェノール/クロロホルム
- エタノール
- TE
- PCR試薬キット
- ポリヌクレオチドキナーゼ
- DNAライゲーション試薬キット
- 制限酵素各種
- コンピテントセル
- LB培地
- LB寒天培地
- SOC培地
- アンピシリン
- プラスミド抽出キット
- RNA合成試薬キット
- 酢酸カリウム溶液
- ホルムアルデヒド
- ホルムアミド
- MOPS
- Transdirect insect cell(島津製作所、292-30000-91)
- FluoroTect TM GreenLys in vitro Translation Labeling System (プロメガ社、L5001)
- Anti-FLAG® M2 Agarose from mouse(SIGMA社、A2220)
- Strep-Tactin® Superflow(QIAGEN社、30001または30003)
- FLAG Peptide(SIGMA社、F3290)
- Desthiobiotin(SIGMA社、D1411-1G)
実験の詳細
Ⅰ:発現プラスミドの構築
5’UTRと開始コドンの距離が長くなるとタンパク質合成量が低下する傾向があるため、開始コドンは出来るだけ5’UTRに近い位置に挿入する。経験的にpTD1のEcoRV/KpnIサイトへのライゲーション効率が最も高いことを確認しており、本プロトコルに従えば、インサートサイズにも依るものの、90%以上のコロニーがインサートの挿入されたクローンとして得られる。そのため、可能な限りEcoRV/KpnIサイトへの挿入をお奨めする。ちなみに、EcoRV/EcoRIサイトに挿入した場合のクローニング効率は20~30%、EcoRV/BamHIでは<10%程度となる。SmaIおよびXbaIについては検討を行っていない。
1.インサートDNAの調製
① プライマー設計
以下の2種類のプライマーを合成する(脱塩グレードで問題なし)。合成タンパク質にアフィニティ精製用のタグを付加する場合は、「Ⅴ:合成タンパク質のアフィニティ精製」を参照のこと。

- 15-30 merで設計し、2×(AとTの数)+4×(GとCの数)=55-60前後となるよう設計する。
- 成熟領域などを発現させる場合においても開始コドンを目的配列の直前に入れるよう設計する。
- PCR産物のリン酸化ステップを省くため、5’末端リン酸化プライマーでも構わない。

- 25-40 merで設計し、ストップコドン以降の配列は2×(AとTの数)+4×(GとCの数)=55-60前後となるよう設計する。
- 制限酵素サイトの前に2-3塩基の付加配列を設ける。KpnIの場合はGGの2塩基を付加する。その他の制限酵素については、各メーカーからの情報を参照のこと。
- 挿入する制限酵素サイトとしては目的遺伝子内にその制限酵素サイトが無いものを選択する。
- Stopコドンに関しては、目的遺伝子に対応するコドンで構わない。
② 目的遺伝子の増幅
PCRは、Terminal transferase活性が無いFidelityの高い酵素を使用する。PCR反応後の反応液2μLをアガロースゲル電気泳動に供し、目的サイズの位置にバンドがあることを確認する。その後、フェノール/クロロホルム抽出、エタノール沈殿により増幅産物を精製する。また、各社より市販されている精製キットを使用しても構わない。
以下にKOD plus(TOYOBO社、KOD-201)を用いた場合の反応条件を記す。

③ 5’末端のリン酸化と制限酵素処理
PCR産物の5’末端をリン酸化する。リン酸化後、エタノール沈殿により精製し、制限酵素処理を行う。以下にTOYOBO社のT4 Polynucleotide Kinase(PNK-111)を用いた場合の反応条件を示す。
まず、エタノール沈殿後の沈殿を35 μL の Denaturation Buffer(TOYOBO 社 T4 Polynucleotide Kinase に付属)に溶解する。90℃、2分間の熱処理後、氷中で急冷し、リン酸化反応を行う。
| DNA溶液 | 35 μL |
| Blunt End Kinase Buffer | 5 μL |
| 0.1M ATP | 0.5 μL |
| T4 Polynucleotide Kinase | 1 μL |
| 滅菌蒸留水 | 8.5 μL |
| 50 μL |
37℃、1時間反応後、反応液を90℃で2分間処理し、室温になるまでゆっくり冷ます。その後、エタノール沈殿により精製する。次に、沈殿を85μLの滅菌水に溶解し、C末端プライマーに導入した制限酵素で消化する。反応条件の一例を以下に示す。
DNA溶液 85 μL 10×Buffer 10 μL 制限酵素 30-50 U 滅菌蒸留水 to 100 μL
37℃、2時間反応後、フェノール精製や市販の精製キットを用いて精製し、一部を分光光度計にて定量する。
2. 発現ベクター(pTD1)の調製
ここでは、EcoRV/KpnIサイトへのライゲーションを例に、発現ベクターの調製法について示す。
まず、5μgのpTD1をEcoRVで消化する。反応条件を以下に示す。
| pTD1 | 5 μg (キット添付のDNAを使用する場合は10 μL) |
| 10×Buffer | 5 μL |
| EcoRV | 30 U |
| 滅菌蒸留水 | to 50 μL |
37℃、1時間で反応後、反応液1μLをアガロースゲル電気泳動に供し、約3kbpの位置にバンドがあることを確認する。その後、エタノール沈殿により精製する[工夫とコツ1]。
沈殿を85μLの滅菌水に溶解し、もう一方の制限酵素で消化する。反応条件を以下に示す。
| DNA溶液 | 85 μL |
| 10×Buffer | 10 μL |
| 制限酵素 | 30-50 U |
| 滅菌蒸留水 | to 100 μL |
37℃、2時間で反応後、フェノール精製や市販の精製キットを用いて精製し、一部を分光光度計にて定量する。この結果、約3.5μg程度のベクターが得られる。
3. ライゲーション
1および2で作製したインサートとベクターをライゲーションする。ここではNEW ENGLAND Biolabs社のQuick LigationTM Kit(M2200S)を用いたライゲーション反応の例を示す。
| ベクター(10 μg/mL) | 2 μL |
| インサート | (ベクター:インサート)のモル比が(1:10)となるよう添加 |
| 2×Quick Ligation Reaction Buffer | 10 μL |
| Quick T4 DNA Ligase | 1 μL |
| 滅菌蒸留水 | to 20 μL |
25℃、5分間反応する。
4. 形質転換
3.で調製したライゲ-ション反応液を用いて大腸菌を形質転換する。使用するコンピテントセルには、DH5αを推奨する。ここでは市販のE. coli DH5α Competent Cells(タカラバイオ、9057)を用いた例を示す。
2μLのライゲーション反応液にCompetent Cellsを20μL添加し、氷上で30分間静置する。次に42℃で1分間熱処理し、再度氷上で5分間静置する。SOC培地を180μL添加し37℃で1時間インキュベートした後、培養液100μL~全量をLBアンピシリンプレートにストリークし、37℃で終夜培養する。
以上の操作で数十~数百の形質転換体が得られる。pTD1は青白判別に対応していないため、以下の方法によってインサートが挿入されたことを確認する。
5. インサートチェック
目的遺伝子が挿入されたかどうかは、プラスミド抽出物またはコロニーからのPCRにより確認する。EcoRV/KpnIサイトに目的遺伝子を導入した場合は、ほとんどが目的遺伝子を有する形質転換体として得られるため、プラスミド抽出を推奨する。その他の制限酵素サイトに導入した場合は、コロニーからのPCRによりインサートチェックを行うことを推奨する。
①プラスミド抽出
コロニーをLBアンピシリン培地(1.5mL)に植菌し、37℃で終夜振とう培養する。培養液からGenEluteTM PlasmidMiniprep Kit(SIGMA社、PLN-70)などを用いてプラスミドを抽出する。精製したプラスミド溶液5μL分をアガロースゲル電気泳動に供し、未切断のpTD1ベクターとの移動度を比較し、インサートの有無を確認する。
②コロニーPCR
ここではTOYOBO社InsertCheck-Ready-Blue(PIK-251)を用いたインサートチェックの例を示す。
増幅用のプライマーとしては下記のものを使用する。
pTD1-161-179: 5’-GCAGATTGTACTGAGAGTG-3’
pTD1-845-827: 5’-GGAAACAGCTATGACCATG-3’
InsertCheck-Ready-Blue 1 mLに対し、上記プライマーをそれぞれ150 pmolずつ添加し、PCR混合液を作製する。PCR混合液を10μLずつPCR用のチューブに分注し、コロニーをチップ(爪楊枝でも可)で突付き、レプリカ用のLBアンピシリンプレートに植菌する。続いてPCR混合液にチップを付けてすすぎ、PCRを行う。

反応液を全量、アガロースゲル電気泳動に供し、インサートのサイズ(kbp)+0.7kbpの位置にバンドがあることを確認する。インサートが挿入されていないものは約0.7kbpの位置に泳動される。
6. プラスミド抽出
インサートの挿入が確認された形質転換体について、LBアンピシリン培地に植菌し、プラスミド抽出を行う。この段階では、後のmRNA合成用の鋳型として使用するため、各社プラスミド抽出キットの使用を推奨する。1.5mLの培養液あたり5μg以上のプラスミドが取得可能であるので、mRNAの合成スケールに応じて培養量を選択する。DNA抽出キットを用い、インサートチェックを行った場合、本ステップは必要ない。
7. シークエンス
本プロトコルに従いPCR産物をpTD1ベクターに挿入した場合、20%程度のクローンに開始コドンのAが欠落した変異が認められる。また、プライマー部位にも変異が認められるケースがあるため、最低でも下記2種のプライマーを用いてシークエンスの確認を行う必要がある。また、極稀に内部配列にも変異が認められる場合もあるため、全長シークエンスの確認を推奨する。
シークエンス用のプライマーとしては下記に示す、pTD1-161-179(N末端側プライマー)、pTD1-412-394(C末端側プライマー)の使用を推奨する。アニーリング温度は50℃である。
pTD1-161-179: 5’-GCAGATTGTACTGAGAGTG-3’
pTD1-412-394: 5’-ACAACGCACAGAATCTAGC-3’
8. mRNA合成用鋳型の調製
mRNA合成用鋳型は、プラスミドを制限酵素により直鎖化、またはPCRにより調製する。いずれの方法を選択しても構わない(PCRで行う場合はFidelityの高い酵素を使用する)。
① 制限酵素による直鎖化
プラスミドをHindⅢまたはNotIで消化する。目的遺伝子中に、これらの制限酵素切断サイトがある場合には、Transdirect取扱説明書7ページ記載の酵素を用いて直鎖化する。制限酵素処理後、RNaseのコンタミネーションを防ぐために、必ずフェノール/クロロホルム抽出を行う[工夫とコツ2]。その後エタノール沈殿により精製し、少量の滅菌水に溶解する(125μg/mL以上となるよう注意する)。
② PCRによる鋳型の作製
下記条件にてPCRを行う。ここではFidelityの高いKOD plusを用いた場合の反応条件を示す。

反応液2μLをアガロースゲル電気泳動に供し、目的サイズ(インサートのサイズ(kbp)+0.7kbp)の位置にバンドがあることを確認する。次に、フェノール/クロロホルム抽出、エタノール沈殿により増幅産物を精製する。精製後、少量の滅菌水またはTEに溶解し(50μLの反応液あたり20μL程度で溶解する)、その一部を分光光度計にて定量する。50μLの反応液あたり5μg以上のPCR産物が取得可能である。
Ⅱ:mRNAの合成と精製
mRNAの合成には市販のRNA大量合成キットを使用する[工夫とコツ3]。また、mRNA精製の目的は、未反応NTPと塩の除去である。未反応のNTPは、分光光度計を用いたmRNA濃度定量時に検出されるため正確な定量を妨げる(見かけ上、mRNA量が増加)。タンパク質合成時のmRNA濃度は非常に重要であるため、mRNAの正しい定量が必要となる。そのため、ゲル濾過カラムによる未反応NTPの除去を推奨する。
1. mRNAの合成
ここでは、反応時間が短く使い勝手の良い、下記の2つのキットを用いたmRNA合成例を示す。 タンパク質合成反応液量に従って、スケールアップ・ダウンして合成する。100μL反応スケールで合成した場合、その後の精製を経て、およそ300-600μg程度のmRNAが取得可能である。
①Epicentre社 AmpliScribe™ T7-Flash™ Transcription Kit (ASF3257)
| 鋳型DNA | 5 μg |
| 10×Buffer | 10 μL |
| ATP | 9 μL |
| CTP | 9 μL |
| GTP | 9 μL |
| UTP | 9 μL |
| DTT | 10 μL |
| T7 RNA Polymerase | 10 μL |
| RNase Free Water | to 100 μL |
反応は、37℃で30分間行う。
②Promega社 T7 RiboMAX™ Express Large Scale RNA Production System (P1320)
| 鋳型DNA | 5 μg |
| 2×Buffer | 50 μL |
| T7 RNA Polymerase | 10 μL |
| RNase Free Water | to 100 μL |
反応は、37℃で30分間行う。
いずれのキットの場合にも、反応終了後は一旦冷却などはせずに、速やかに精製を行うこと。
[工夫とコツ4]
2. mRNA精製
mRNAの精製はゲル濾過による未反応NTP・塩の除去とエタノール沈殿による濃縮を目的に行う。フェノール精製などは特に必要ない。また、使用する水も滅菌蒸留水で問題ない。ここではGE社製NICKTM Columns(17-0855-01)を用いた精製法について示す。
カラムの上フタを外し、溶液を捨てる。3mLの滅菌蒸留水でリンスし、再度滅菌蒸留水を捨てる。カラム先端のキャップを外し、スタンドに立て、ゲルを平衡化するため3mLの滅菌蒸留水を加え、完全に流しきる。mRNA溶液をゲル上端にのせ(最大100μLまで)、完全に流しきる。400μLの滅菌蒸留水を加え、完全に流しきる。mRNA溶液を受けるため、1.5mLチューブをカラムの下に配置し、400μLの滅菌蒸留水を加え、mRNA溶液を回収する。回収したmRNA溶液に40μLの3M 酢酸カリウムと950μLのエタノールを加え、よく混合し15,000rpm、4℃で20分間遠心分離する。上清は捨て、70%エタノールでリンスを行い、mRNAの合成スケールに応じて20~100μLの滅菌蒸留水に溶解する。mRNA濃度は2mg/mL以上に調整すること[工夫とコツ5]。
3. mRNAの確認
mRNAが正しく合成できたかどうかは、精製後に電気泳動で確認する事が可能である。 初めてmRNAを合成、精製したような場合には、精製後に正しくmRNAが合成できたかどうかを電気泳動によって確認することを推奨する。以下にプロトコルと実験結果の一例を示す。
| 20×MOPS | 1 μL |
| ホルムアルデヒド | 3 μL |
| ホルムアミド | 8 μL |
| サンプル溶液 | 8 μg |
| 滅菌蒸留水 | to 20 μL |
上記の組成にて混合後、65℃で15分間加熱処理する。1%アガロースゲルにて電気泳動し、エチジウムブロマイドにて検出する。 結果を図1に示す。図のようにバンドが検出されれば問題ない。逆に、「バンドが見えない」もしくは「スメアになっている」場合は、mRNAの分解が考えられるため、実験過程でRNaseのコンタミがなかったか、鋳型DNA調製時にフェノール/クロロホルム抽出を行ったかなど、確認する。
Ⅲ:タンパク質合成
TransdirectキットからReaction Buffer、4 mM Methionine、Insect Cell Extractを取り出し、氷上にて溶解する。また、これ以降の工程は、タンパク質合成を開始するまで氷上で行う[工夫とコツ6]。 下記に示す組成にて、反応液(50μLスケール)を調製する[工夫とコツ7]。
Reaction Buffer 15 μL 4 mM Methionine 1 μL Insect Cell Extract 25 μL mRNA 16 μg 滅菌蒸留水 to 50 μL 穏やかに混合した後、必要であればスピンダウンして25℃で5時間インキュベートする。反応終了後は速やかに氷上に移し、その後の実験に用いる。
Ⅳ:タンパク質の発現確認
通常、SDS-PAGE後のCBB染色による目的タンパク質の発現確認は難しい。そこで、Transdirectを用いたタンパク質の発現は、蛍光試薬によるラベル、RIによるラベル、ウェスタンブロッティングなどで確認する。ここでは、非常に簡便に発現を確認することが出来る、蛍光試薬によるラベル化法について示す。蛍光試薬としてはプロメガ社FluoroTect TM GreenLys in vitro Translation Labeling System (L5001)を用いる。50μLあたり1μLのFluoroTectを反応液に加え、反応を行う。以下にその組成を示す。
| Reaction Buffer | 15 μL |
| 4 mM Methionine | 1 μL |
| Insect Cell Extract | 25 μL |
| FluoroTect | 1 μL |
| mRNA | 16 μg |
| 滅菌蒸留水 | to 50 μL |
穏やかに混合した後、必要であればスピンダウンして25℃で5時間インキュベートする。反応終了後、2μLのSDS-PAGE loading buffer (4X)を反応液6μLに対して添加し、70℃で3分間熱処理する。このサンプルをSDS-PAGEで分離後、レーザーベースの蛍光イメージアナライザーにて検出する[工夫とコツ8]。実験例として、β-ガラクトシダーゼを発現した結果を図2に示す。
Ⅴ:合成タンパク質のアフィニティ精製
N末端、或いはC末端にstrep-tag®(IBA GmbH)またはFLAG®-tag(Sigma-Aldrich Corp.)を導入することで、簡便に目的タンパク質を回収することが可能である。ここでは、これらのプロトコルを示す[工夫とコツ9]。
1.cDNAへのタグ配列の導入とpTD1へのクローニング
① プライマーの設計
目的に応じて、下記いずれかのプライマーセットを合成する。

② PCRによる鋳型DNAの作成
「Ⅰ:発現ベクターの構築」のプロトコルに従って、目的遺伝子を増幅し、pTD1ベクターに挿入する。
2.mRNAの合成と精製
「Ⅱ:mRNAの合成と精製」に従って、mRNAを調製する。反応スケールは100μLで行う。これにより、およそ500μg程度のmRNAが取得可能である。
3.タンパク質合成
「Ⅲ:タンパク質合成」に従って、目的タンパク質を合成する。
1 mLスケールにてタンパク質合成を行う。下記に組成を示す。
| mRNA | 320 μg |
| 4 mM Methionine | 20 μL |
| Reaction Buffer | 300 μL |
| Insect Cell Extract | 500 μL |
| 滅菌蒸留水 | to 1000 μL |
穏やかに混合した後、必要であればスピンダウンして25℃で5時間インキュベートする。反応終了後、15,000 rpmにて15分間遠心分離を行う。
4. アフィニティ精製
mRNA精製に使用したNick™ columnsを洗浄した空きカラムを用いてアフィニティカラムを作製する。カラムの作製はある程度の時間を要するため、タンパク質合成を行う前に行うことを推奨する。
①strep-tag®の場合
Strep-Tactin® Superflow(QIAGEN社30001または30003)のオープンカラムを作製する。50%のスラリーとして供給されるので、カラム1本あたり1 mLのスラリーを使用する。mRNA精製に使用したNickTM columnsを洗浄した空きカラムに上述のスラリーを充填し、フィルターをカラム上端に設置する。以上の操作でbed volume 0.5mLのオープンカラムが作製できる。以下に精製の手順を示す。
50mM Tris-HCl, pH8.0, 300mM NaCl(Buffer A、5mL)でカラムを平衡する。カラムにタンパク質合成反応液の遠心上清をアプライする。0.5mLのBuffer Aでカラムを洗浄する。この操作は5回繰り返す。2.5mM Desthiobiotin(SIGMA社、D1411-1G)を含むBuffer Aを1.5mL添加し溶出する。溶出液をスピンタイプの限外ろ過で20-50μL程度まで濃縮する[工夫とコツ10]。
②FLAG®-tagの場合
Anti-FLAG® M2 Agarose from mouse(SIGMA社、A2220)を用いて、上記と同様の操作でオープンカラムを作製する。 50%のスラリーとして供給されるので、カラム1本あたり1 mLのスラリーを使用する。以下に精製の手順を示す。
はじめに、タンパク質合成反応液にはDTTが含まれるため、反応液を50mM Tris-HCl, pH8.0, 150mM NaCl(Buffer B)で平衡化したPD-10(GE社、17-0851-01)にアプライし脱塩する。 次に、作製したカラムをBuffer B 5mLで平衡化する。カラムに脱塩処理を行ったPD-10の溶出液をアプライする。1mLのBuffer Bでカラムを洗浄する。この操作は5回繰り返す。100μg/ mL FLAG Peptide(SIGMA社、F3290)を含むBuffer Bを2.5 mL添加し、溶出する。溶出液をスピンタイプの限外ろ過で20-50μL程度まで濃縮する[工夫とコツ11]。
実験例として、β-ガラクトシダーゼのN末端、C末端にそれぞれstrep-tag®またはFLAG®-tagを導入し、アフィニティ精製を行った結果を図3に示す。典型的な例として、このように 1 mLの反応液あたり10-20μgのタンパク質が取得可能である。しかしながら、どちらのタグを用いるか、またN末端C末端どちらにタグを導入するかは、タンパク質の種類によって大きく異なる。次項にタンパク質精製時におけるトラブルシューティングおよびその対処法を記す。
5.精製に関するトラブルシューティング
○ 目的タンパク質の収量が少ない
- 目的タンパク質が可溶性画分にあることを確認する。可溶性画分にある場合、カラムに吸着していないことが考えられる。このような場合は、使用するタグの種類、タグの導入位置を検討する。その際に、次項で詳述するようなスペーサー配列の目的遺伝子とタグ配列の間への挿入についても検討する。
- 排除分子量の小さな限外ろ過膜を用いて濃縮する。
- カラムを新しいものに変更する。
○ 目的タンパク質の精製度が低い
- 抽出液由来タンパク質のカラムへの非特異的吸着、または目的タンパク質への吸着が考えられる。このような場合、バッファー系を検討する。Strep-Tactin® Superflowでは、1% Triton X-100、1% Tween、0.3% CHAPS、2% Igepal CA-630、1M NaClなどが使用可能である。Anti-FLAG® M2 Agarose from mouseでは、5% Triton X-100、5% Tween20、0.1% CHAPS、0.1% Igepal CA-630、1 M NaClなどが使用可能である(詳細は各製品の取扱説明書参照)。
- 膜貫通領域やシグナルペプチドなどの疎水性領域を含むタンパク質を合成、精製した場合、約50kDa、70kDa付近の抽出液由来のタンパク質がコンタミするケースが多く観察されている(50kDaのタンパク質についてはβ-チューブリンであることを確認済み)。このような場合は疎水性領域を欠損させたコンストラクトを構築することを推奨する(例:シグナルペプチドを有するタンパク質を成熟型タンパク質として発現)。
6.スペーサー配列を含むタグ発現用ベクターの構築
目的遺伝子とタグ配列の間にスペーサー配列を挿入することで、回収率が飛躍的に改善される場合がある。ここでは、C末端タグの前に8つのグリシン残基からなるスペーサー配列が挿入される発現ベクターの構築法を記す。
①strep-tag®の場合
以下のプライマーを合成する。
G8-strep-F: 5’-GGGAATTCGGTACCGGATCCGGTGGAGGTGGAGGTGGAGGTGGATGGAGCCATCCGCAGTTTGAAAAGTAATCTAGAGC-3’
G8-strep-R: 5’-GCTCTAGATTACTTTTCAAACTGCGGATGGCTCCATCCACCTCCACCTCCACCTCCACCGGATCCGGTACCGAATTCCC-3’ 両者のプライマーを混合しアニールする。EcoRIおよびXbaIで消化し、これをpTD1のEcoRI-XbaIサイトに挿入する。
②FLAG®-tagの場合
以下のプライマーを合成する。
G8-FLAG-F: 5’-GGGAATTCGGTACCGGATCCGGTGGAGGTGGAGGTGGAGGTGGAGACTACAAGGATGACGATGACAAGTAATCTAGAGC-3’
G8-FLAG-R: 5’-GCTCTAGATTACTTGTCATCGTCATCCTTGTAGTCTCCACCTCCACCTCCACCTCCACCGGATCCGGTACCGAATTCCC-3’ ①と同様の操作でpTD1のEcoRI-XbaIサイトに挿入する。
構築したベクターの塩基配列を以下に示す。

本ベクターへの目的遺伝子のクローニングにおいて、N末端側プライマーは開始コドン以下の配列を使用する。C末端側のプライマーには、ベクターに既に組み込まれているためストップコドンを含めない。また、C末端プライマーにはEcoRI、KpnI、BamHIサイトに挿入できる制限酵素サイトを導入する。開始コドンは、EcoRVの切断サイトに挿入することを推奨する。
工夫とコツ
- EcoRVとバッファーが同一でも高いライゲーション効率を得るために、一度エタノール沈殿で精製した後、もう一方の制限酵素消化を行うことをお奨めする。
- プラスミドの抽出にキットを用いた場合、その際に使用されるRNase Aも僅かながら混入してくる。この僅かな混入がmRNA合成に大きく影響することがあるため、フェノール/クロロホルム抽出はこのステップにおいて必ず行うこと。
- 推奨キットについてはTransdirect取説8ページ参照。
- いずれのキットもバッファーが溶解しにくい場合がある。このような場合、完全に溶解させないと著しく反応効率が低下する。溶けにくい場合は、60℃で数分加温すると速やかに溶解するため、必ず完全に溶解させてから使用すること。また、反応時間は厳守すること。特に鋳型のサイズが大きい場合、過剰な反応を行うと、反応中に沈殿を生じる事がある。沈殿が生じてしまうと、mRNAは回収できない。このような場合は、①反応時間を20分程度まで短縮する、②鋳型DNAの量を2-3割程度減らす、③鋳型を制限酵素処理ではなく、PCRにより調製するなどで改善が見られる。
- エタノール沈殿時に乾固させてしまうと、mRNAが水に溶解できなくなる。そのため、mRNAは乾固させず、70%エタノールでのリンス後は、ピペットで余分なエタノールを除去する程度で構わない。
- Reaction Bufferと4 mM Methionineは、室温で溶解しても構わない。使用後の各試薬は速やかにフリーザーへと戻す。Insect Cell Extractの凍結融解は、8回程度までその性能に影響がないことを確認している。
- 反応スケールは目的に応じて自由に変えることが可能であり、スケールアップまたはダウンによる合成効率の低下は認められていない。また、RNaseインヒビターを反応系に最終濃度1unit/μLで添加することにより、タンパク質合成量が改善されることがある。
- 本手法では、20kDa付近に未反応のFluoroTect試薬である、蛍光標識リジン-tRNAが検出されるため、目的タンパク質が20kDa以下と予想される場合は、反応終了後の反応液をRNase Aで処理すると、容易に目的タンパク質が検出できる。
- 目的のタンパク質によっては、後に示すようなスペーサー配列の導入が必要な場合もあるため、まずは精製を行う前に、「Ⅳ:タンパク質の発現確認」で示したような発現確認を行うとトラブルが生じた時に対処しやすい。
- 溶出液にDesthiobiotinを用いた場合、カラムの再生が可能である。再生法については、Strep-Tactin® Superflow添付の取扱説明書を参照すること。5-10回程度であれば、極端な収率の低下は認められていない。
- カラムの再生が可能である。再生法については、Anti-FLAG® M2 Agarose from mouse添付の取扱説明書を参照すること。5-10回程度であれば、極端な収率の低下は認められていない。
参考文献
- Ezure, T., Suzuki, T., Higashide, S., Shintani, E., Endo, K., Kobayashi, S., Shikata, M., Ito, M., Tanimizu, K., and Nishimura, O. (2006) Cell-free protein synthesis system prepared from insect cells by freeze-thawing. Biotechnol. Prog. 22, 1570-1577.
- Suzuki, T., Ito, M., Ezure, T., Kobayashi, S., Shikata, M., Tanimizu, K., and Nishimura, O. (2006) Performance of expression vector, pTD1, in insect cell-free translation system. J. Biosci. Bioeng. 102, 69-71.
- Suzuki, T., Ito, M., Ezure, T., Shikata, M., Ando, E., Utsumi, T., Tsunasawa, S., and Nishimura, O. (2006) N-Terminal protein modifications in an insect cell-free protein synthesis system and their identification by mass spectrometry. Proteomics 6, 4486-4495.
- Suzuki, T., Ito, M., Ezure, T., Shikata, M., Ando, E., Utsumi, T., Tsunasawa, S., and Nishimura, O. (2007) Protein prenylation in an insect cell-free protein synthesis system and identification of products by mass spectrometry. Proteomics 7, 1942-1950.
-

図1: -
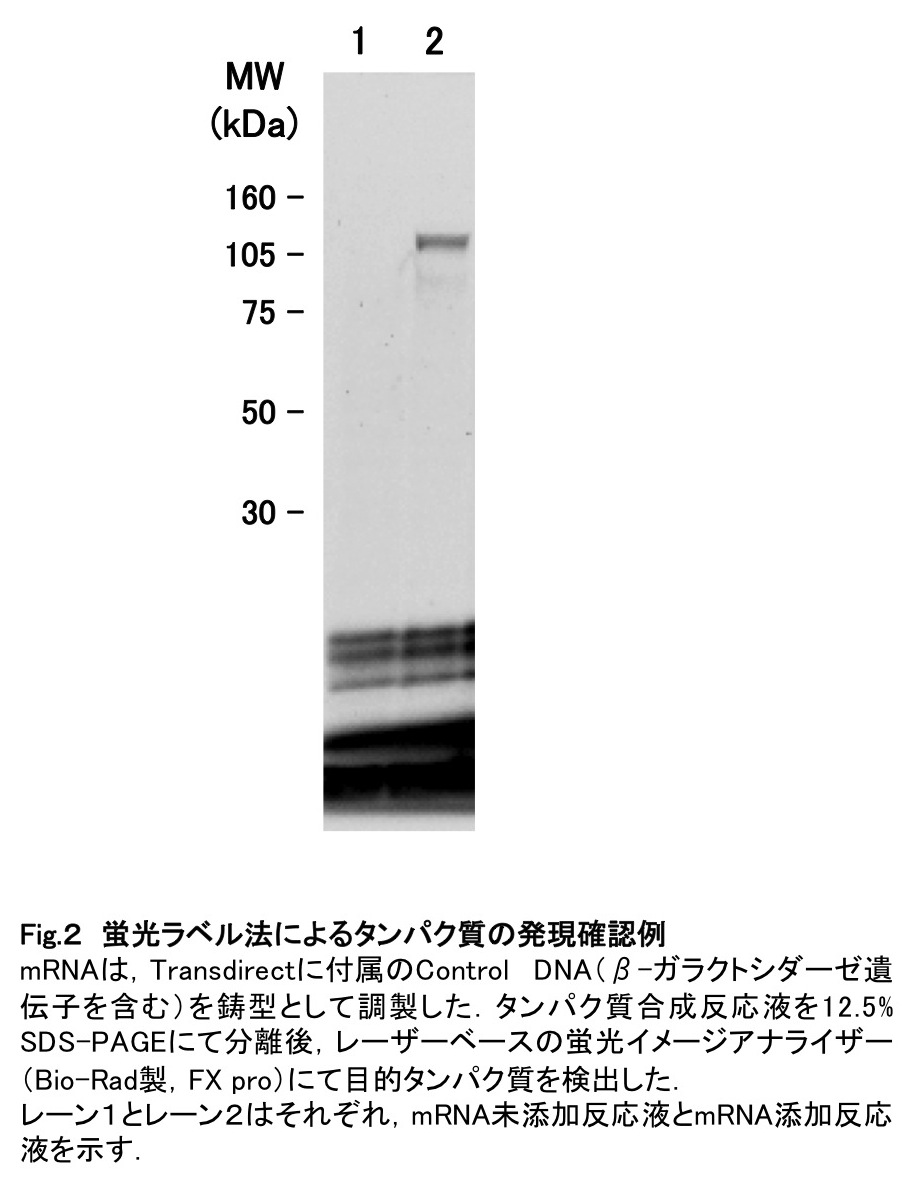
図2: -

図3:
