

概要
安定同位体標識した組換え蛋白質の大量発現においては、ハンドリングの手軽さ、増殖の速さ、標識コストの低さ等の理由から、大腸菌発現系が最も広く用いられている。しかし、原核細胞である大腸菌を用いた発現系では、高分子量蛋白質、膜蛋白質、複雑なジスルフィド結合を含む蛋白質、高等生物由来蛋白質等の発現量が不十分になることも多い。
酵母細胞はもっとも単純な真核細胞であり、その培養は大腸菌と同様の手軽さで行えるうえに、上記のように大腸菌での試料調製が困難な蛋白質を発現させる際には、より強力なオプションとなる。また近年、酵母発現系そのものの技術的な発展に加え、酵母発現系を用いた様々な安定同位体標識法やアプリケーションが開発されており、その有用性と汎用性は年々向上している。このように、酵母発現系は NMR を用いた蛋白質立体構造解析を指向した発現系として、大腸菌発現系とならぶ現実的な選択肢の一つとなりつつある。
本稿では、特に酵母Kluyveromyces lactis(K. lactis)を宿主として用いた安定同位体標識蛋白質の大量発現について、方法の概要から実際に実験を行ううえでのコツに至るまで広く概説する。
装置・器具・試薬
- 恒温培養器(30℃および37℃)
- 恒温振とう培養器(30℃,250 rpmでのレシプロ培養が可能なもの)
- 三角フラスコ(500 mL容量)
- メジュームビン
- 濾過滅菌用フィルターユニット
- 空気ポンプ(ホームセンターで市販されている小型の魚類飼育用のもので十分)
- ジャーファーメンター培養装置
- ペリスタポンプ(数mL/hの低速域でも安定して稼働するものが好ましい)
- 塩化カルシウム管(空のエコノパックカラム等に塩化カルシウム顆粒を詰めたものでも良い)
- 純水調製装置
- オートクレーブ滅菌装置
- pKLACプラスミドDNA(NEB)
- K. lactis GG799株(NEB)
- K. lactis protein expression kit(NEB)
- 制限酵素類
紹介する実験手順
- 酵母K. lactis発現系の構築
- 酵母K. lactisの形質転換・形質転換体の選択
- 均一 \(\ce{^{13}C,^{15}N}\) 標識蛋白質の発現
- 均一 \(\ce{^{2}H}\) 標識を伴う安定同位体(均一 \(\ce{^{15}N}\))標識法
- 均一 \(\ce{^{2}H}\) 標識を伴う安定同位体(ILVメチル基選択的 \(\ce{^{1}H,^{13}C}\))標識法
実験の詳細
1)酵母K. lactis発現系の構築
酵母K. lactisで目的蛋白質を発現させるためのプラスミドベクターおよび宿主酵母細胞株(コンピテント化済み)は、New England Biolabs(NEB)から「K. lactis Protein Expression Kit」として市販されている。
酵母発現系では、目的蛋白質のN末端にK. lactis由来α-Mating factorのような分泌シグナル配列を付加することにより、目的蛋白質を細胞外(培地中)へ分泌発現させることが可能である。細胞外(培地中)へ目的蛋白質を発現させる戦略は、a)酵母細胞質に存在する強力なプロテアーゼによる目的蛋白質の非特異的デグラデーションを回避できる、b)酵母の分泌経路を通る過程で目的蛋白質の立体構造形成(特に、適切なジスルフィド結合の形成)が促され、正しい立体構造および生理活性を保持した目的蛋白質が得られる、c)培地中は夾雑分子の量が細胞破砕液に比べて少なく、細胞破砕液から精製する場合に比べて目的蛋白質の精製が容易になる等のメリットがある。
NEBから現在入手可能なK. lactis発現ベクターpKLAC2にもK. lactis由来α-Mating factor配列が備わっており、適切な制限酵素サイトに目的蛋白質のcDNA(目的蛋白質のN末端にLys-Argが付加するようにcDNA配列を設計する必要がある)を挿入することにより、目的蛋白質を培地中に分泌発現させるベクターを作製することが容易にできる。このようにして構築されたベクターから発現する蛋白質は、分泌時に、酵母のゴルジ体に存在するKexプロテアーゼによってLys-Argと目的蛋白質の間が切断され、付加配列のない目的蛋白質のみが培地中に分泌される。
目的プラスミドの構築についての詳細は、NEBの取扱説明書を参照いただきたい。
2)酵母K. lactisの形質転換・形質転換体の選択
目的蛋白質をコードするcDNAをもつpKLACベクターを、制限酵素Sac II(認識配列 CCGCGG)もしくはBst XI(認識配列 CCANNNNNNTGG,Nは任意の塩基)で切断し、直鎖型にしたものをK. lactis GG799株に遺伝子導入する。(そのため、目的蛋白質をコードするcDNAの配列内にSac IIもしくはBst XIの認識配列が存在しないようにcDNA配列を設計しておく必要がある。)ベクター遺伝子はK. lactisのゲノム中に相同組換えによって挿入され、目的蛋白質を恒常的に発現する形質転換株となる。実際の形質転換手順の詳細は、NEBの取扱説明書を参照いただきたいが、ここでは、K. lactis発現系の形質転換体コロニーのセレクションの戦略について以下概説する。
形質転換体は、アンモニウム塩のような窒素源を含まず、その代わりにアセトアミドを添加した寒天培地上にてセレクションする。通常のK. lactis GG799株は、窒素源の無い寒天培地上では生育できず、コロニーは形成できない。しかし、pKLACベクターはfungal acetoamidase(amdS)遺伝子も有しており、目的遺伝子とともにamdS遺伝子もK. lactisのゲノムに組み込まれる。その形質転換体はacetoamidaseを使ってアセトアミドをアンモニウムに代謝分解することができるため、アセトアミドを添加した寒天培地上で生育できるようになる。
この方法は、a)メタノール資化性酵母Pichia pastoris(P. pastoris発現系のように、形質転換体のセレクションに高価な抗生物質(Zeocin等)を大量に使用する方法に比べてコストを抑えられる、b)pKLACベクターがK. lactisゲノムに組み込まれたコピー数が多い株ほど、そのアセトアミド含有寒天培地上での生育効率が高まるため、生育の良い(大きな)コロニーを選ぶことにより、目的遺伝子が複数個ゲノムに組み込まれた(マルチコピー)株を選択取得しやすい、等の利点がある。
特にb)の性質は、目的遺伝子がマルチコピーでゲノムに挿入され、目的蛋白質の発現量が高い形質転換体を取得することが期待されるだけでなく、異なる目的蛋白質をコードする複数種類のベクターを同時にトランスフォームすれば、複数種類の目的蛋白質を共発現する形質転換体を得る事も容易である。筆者の経験的には、生育の良いコロニーが20個形成されたとすると、マルチコピー株(もしくは共発現株)はその中にほぼ確実に含まれている。一方、P. pastoris発現系のように抗生物質で形質転換体をセレクションする方法の場合、マルチコピー株を発生させるには、抗生物質の濃度が高い寒天培地でコロニー形成させることによりその確率を上昇させるのが一般的である。しかし、筆者自身が感じた限りでは、抗生物質の濃度とマルチコピー株発生の相関は直線的ではなく、抗生物質の濃度が高い寒天培地でセレクションしたからといって必ずしもマルチコピー株が得られるとは限らない。
形質転換体に含まれる目的遺伝子カセットのコピー数は、colony-direct PCRにより定量することが可能である。その具体的な手順は取扱説明書を参照していただきたい。
また、各コロニーの目的蛋白質の発現量は、小スケール(培地量2 mL程度)での培養とSDS-PAGEにより確認できる。その手順の詳細はNEBの取扱説明書を参照していただきたいが、分泌発現の場合、形質転換体コロニーを2 mLのYPD培地にて培養(30℃でレシプロ振とう培養(250 rpm)、2~3日間)した後、培地上清を100 μLほど回収して、菌体を遠沈し、その上清10 μLをSDS-PAGEに供することで、ゲルのCBB染色で発現量を目視確認できる。また、これで目視確認できるほどの発現量があるコロニーは、後述の手順で目的蛋白質を大量発現できる可能性が高い。できるだけ発現量の高いコロニーを取得・グリセロールストックするようにする。このとき次節で説明するように、蛋白質の均一 \(\ce{^{13}C}\) 標識を行う場合、\(\ce{^{13}C}\) 標識グルコースを炭素源として培養・蛋白質発現誘導を行うため、YPGal培地ではなくYPD培地を使って蛋白質発現量を確認し、最も発現量の多い形質転換体を選択する。
目的蛋白質を高発現する形質転換株は、グリセロールストックとして−80℃にて長期保存が可能である(NEB取扱説明書を参照)。後述するが、グリセロールストックから培養を開始する場合は、菌体を少量搔き取って YCB–アセトアミド寒天培地に画線し、30℃にて1~2日間静置培養して形成したfreshなコロニーを培養に用いる。
その後のステップである「目的蛋白質の発現のための形質転換株の培養」においては、培地中にアセトアミドが含まれている必要は無い。よって筆者らは窒素源としてアセトアミドではなく塩化アンモニウムを使用している。
YPD培地は下記のように調製する。
- Yeast Peptone(YP)培地
- 20 gのBacto Peptone(BD)と10 gのYeast Extractを950 mLのイオン交換水(超純水でも可)で溶解し、オートクレーブ滅菌。
- 40% [w/v] グルコース溶液
- 200 gのD-グルコースをイオン交換水に溶解して液量500 mLにする。クリーンベンチ内で、オートクレーブ滅菌した空のメジュームビンに濾過滅菌で移す。
- YP+Dextrose(YPD)培地
- クリーンベンチ内で、YP培地 : 40% [w/v] グルコース溶液の液量比が19 : 1となるように混和する(例:YP培地19 mLに40% [w/v] グルコース溶液1 mL添加。必要量のみを用時調製)。
3)均一 \(\ce{^{13}C,^{15}N}\) 標識蛋白質の発現
K. lactisはLAC4プロモーターを目的蛋白質の発現誘導に利用している。LAC4プロモーターの活性はガラクトースによって最も強く誘導され、かつガラクトースはK. lactis自身の生育・増殖にも利用するため、ガラクトースを含む培地で形質転換体を培養するだけで目的蛋白質が恒常的に発現し続ける。
しかし、蛋白質の均一 \(\ce{^{13}C}\) 標識を行う場合、\(\ce{^{13}C}\) 標識ガラクトースは高価(\(\ce{^{13}C}\) 標識グルコースの10–15倍)であり、ガラクトースを炭素源として用いるのは現実的には難しい。K.lactis GG799株の場合、ガラクトースの30–40%程度ではあるが、グルコースでも弱いながらもLAC4プロモーターを誘導する。この点を利用して、均一 \(\ce{^{13}C}\) 標識の場合はガラクトースではなくグルコースを炭素源および蛋白質発現誘導剤として用いることが有効である。
しかし、グルコース含有培地で通常どおり培養したのでは、充分量の蛋白質発現を達成できない。特に安定同位体標識のための最少栄養培地や重水培地などのように、培地の貧栄養性もしくは毒性のために菌体の生育が厳しい条件下での培養では、その傾向がより顕著となる。
そこで筆者らは、ジャーファーメンターを使って菌体の増殖を最大化するとともに、蛋白質発現誘導を恒常的に行うため、数日間にわたって新鮮なグルコース含有培地を連続的に供給し続ける “Fed-batch” 培養法を適用し、グルコースでも1 L培養あたり10 mg以上の目的蛋白質を発現させることに成功した (1–3)。
またこのFed-batch培養法により、蛋白質発現に用いるグルコースの量も5 g/L程度と、大腸菌とほぼ同等の量に抑えることができ、均一 \(\ce{^{13}C}\) 標識にかかるコストを大腸菌発現系と同程度にまで抑えることができる (1–3)。
1. 培地・試薬類の調製
- Yeast Carbon Base(YCB)–アセトアミド寒天培地
- NEBの取扱説明書を参照。
- 1 Mリン酸カリウム緩衝液
- 培養中の培地のpHの変動を抑えるために培地に添加する。目的蛋白質を培地中に分泌発現させる場合、目的蛋白質のpI値を考慮して、目的蛋白質の可溶性に悪影響を与えないようにリン酸緩衝液のpHを決める。培養中はやや酸性に維持したほうが漏出プロテアーゼの活性を最小限に抑えることができる。K. lactisはpH 5.0–8.0の間なら生育・増殖は十分可能である。ちなみに、pH 6.0 ± 0.1の1 Mリン酸カリウム緩衝液を調製するには、132 mLの1 M \(\ce{K2HPO4}\) 溶液と868 mLの1 M \(\ce{KH2PO4}\) 溶液を混和したうえで、水酸化カリウム溶液でpH 6.0に合わせると良い。
- 10× Yeast Nitrogen Base(YNB)溶液
- 34 gのYNB粉末(アミノ酸とアンモニウム塩を含まないもの。べクトンディッキンソン社から入手可能)を1 Lのイオン交換水で溶解する。溶けにくい場合は40–50℃に加温しながら撹拌して溶かす。クリーンベンチ内でろ過滅菌。遮光して4℃保存が可能。
- 500× ビオチン溶液
- 20 mgのビオチン粉末を100 mLのイオン交換水で溶解する。溶けない場合は40–50℃に加温しながら撹拌して溶かす。クリーンベンチ内でろ過滅菌。遮光して4℃保存が可能。
- 200× PTM1溶液
- 下記試薬を1 Lのイオン交換水で溶解させる。\(\ce{CuSO4.5H2O}\) 6.00 g,\(\ce{NaI}\) 0.08 g,\(\ce{MgSO4}\) 3.00 g,\(\ce{Na2MoO4.2H2O}\) 0.20 g,ホウ酸0.02 g,\(\ce{CoCl2}\) 0.50 g,\(\ce{ZnCl2}\) 20.0 g,\(\ce{FeSO4.7H2O}\) 65.0 g,ビオチン0.20 g,\(\ce{H2SO4}\) 5 mL。溶けない場合は40–50℃に加温して溶かす。クリーンベンチ内でろ過滅菌。遮光して4℃保存が可能。
- \(\ce{^{15}N}\) 塩化アンモニウム溶液
- 培養前に用時調製。7.5 gの \(\ce{^{15}N}\) 塩化アンモニウム(98 atom% \(\ce{^{15}N}\))を75 mLのイオン交換水で溶解し、クリーンベンチ内でろ過滅菌。
- D-[\(\ce{^{13}C6}\)]グルコース溶液
- 培養前に必要に応じて用時調製。2 gのD-[\(\ce{^{13}C6}\)]グルコースを50 mLのイオン交換水で溶解し、クリーンベンチ内でろ過滅菌。
- \(\ce{^{13}C,^{15}N}\) 最小培地
- 500 mLの \(\ce{^{13}C,^{15}N}\) 最小培地を、下記の要領で調製する。2 L容量のファーメンタージャーに325 mLのイオン交換水と0.25–0.50 mLの消泡剤Antifoam 204(SIGMA)を入れてオートクレーブ滅菌。室温程度に冷めたら50 mLの10× YNB溶液、1 mLの500× ビオチン溶液、50 mLの1 Mリン酸カリウム緩衝液、2.5 mLの200× PTM1溶液、25 mLの \(\ce{^{15}N}\) 塩化アンモニウム溶液(=終濃度5 g/L)、50 mLの D-[\(\ce{^{13}C6}\)]グルコース溶液(=終濃度4 g/L)を、クリーンベンチ内で無菌的に添加する。
- D-[\(\ce{^{13}C6}\)]グルコース溶液(流加用)
- 培養前に必要に応じて用時調製。6 gのD-[\(\ce{^{13}C6}\)]グルコースを100 mLのイオン交換水で溶解し、クリーンベンチ内でろ過滅菌。
- \(\ce{^{13}C,^{15}N}\) 最小培地(流加用)
- 1 Lの \(\ce{^{13}C,^{15}N}\) 最小培地を、下記の要領で調製する。1 L容量のメジュームビンに650 mLのイオン交換水と0.5–1.0 mLの消泡剤Antifoam 204(SIGMA)を入れてオートクレーブ滅菌。室温程度に冷めたら100 mLの10× YNB溶液、2 mLの500× ビオチン溶液、100 mLの1 Mリン酸カリウム緩衝液、50 mLの \(\ce{^{15}N}\) 塩化アンモニウム溶液(=終濃度5 g/L)、100 mLのD-[\(\ce{^{13}C6}\)]グルコース溶液(流加用)(=終濃度6 g/L)を、クリーンベンチ内で無菌的に添加する。
- 50% [v/v] エタノール溶液
- 適当な容器に水道水200 mLとエタノール200 mLを混和。ジャーファーメンターの排気ホースの先をこの溶液に沈めて、排気の滅菌に用いる(図1)。
2. 培養の手順
- K. lactis形質転換体のシングルコロニーもしくはグリセロールストック少量を滅菌済みの爪楊枝等でピックし、YCB–アセトアミド寒天培地に画線培養する(30℃で1–2日間)。
- Freshなシングルコロニーを、5 mLのYPD培地(50 mLコーニングチューブ)に植菌し、30℃でレシプロ振とう培養(200–250 rpm)、2日間。
- 室温にて3,000 g、5分間の遠心で菌体を遠沈し、2 Lジャー内から \(\ce{^{13}C,^{15}N}\) 最小培地を数十mLとって、それで菌体を穏やかに再懸濁し、2 Lジャー内の \(\ce{^{13}C,^{15}N}\) 最小培地に戻す。
- ポアサイズ0.22 μmのシリンジフィルターを通して滅菌した大気を0.1–0.3 L/minで培地中に供給しながら、30℃で600–800 rpmで撹拌培養。
- ジャーと流加用 \(\ce{^{13}C,^{15}N}\) 最小培地が入ったメジュームビンを、シリコンチューブを介して接続する。そのシリコンチューブにペリスタポンプを接続する(図1)。一定の流速で流加用 \(\ce{^{13}C,^{15}N}\) 最小培地をジャー内の培養液に供給する。例えば流速を約8 mL/hに設定すれば、流加用 \(\ce{^{13}C,^{15}N}\) 最小培地600 mLを75時間(約3日間)かけて培養中に供給し続けることができる。培養期間は2–4日間が一般的であるが、最も収量の高い培養期間は目的蛋白質によって異なるため、小スケール培養による発現量確認の検討(前項参照)で、最も発現量もしくは収量の高くなる培養時間をあらかじめ調べておく。
- 培養終了後、ジャーから培養液を回収し、10,000 gで15分間の遠心により菌体を沈殿させる。目的蛋白質を細胞内に発現させた場合は、遠心上清は廃棄して遠沈した菌体を回収し、菌体を破砕して目的蛋白質の精製へと進む。目的蛋白質を培地中に分泌発現させた場合は、遠心上清(培養後の培地)をポアサイズ0.45 μmのボトルトップフィルターでろ過して、菌体等の夾雑物を除去したうえで培地のみ回収する。その培地から目的蛋白質を精製するまでの間、短期間であれば4℃で保存が可能であるが、その場合はプロテアーゼインヒビターや終濃度1 mM程度のEDTAを添加しておくことを勧める(目的蛋白質の精製においてそのような成分が邪魔になる場合は添加を控え、できるだけすみやかに目的蛋白質の粗精製を済ませる)。
4)均一 \(\ce{^{2}H}\) 標識を伴う安定同位体(均一 \(\ce{^{2}H,^{15}N}\) もしくはILVメチル基選択的 \(\ce{^{1}H,^{13}C}\))標識法
目的蛋白質が高分子量もしくは膜蛋白質等である場合、特定の部位、特にメチル基や芳香族側鎖や特定のアミノ酸の主鎖アミド領域のみを、\(\ce{^{1}H}\) と \(\ce{^{13}C}\) もしくは \(\ce{^{15}N}\) で標識し、それ以外の非易動性 \(\ce{^{1}H}\) を \(\ce{^{2}H}\) で置換することにより、蛋白質内 \(\ce{^{1}H}\) 間の双極子–双極子相互作用による磁化の緩和によるNMRシグナルの広幅化を抑えるとともに、TROSY効果を最大限に得てNMRシグナルの線幅をできる限りシャープに検出する試みを行う。目的蛋白質の \(\ce{^{2}H}\) 置換は、重水(\(\ce{^{2}H2O}\))で調製した培地中にて宿主細胞を培養して目的蛋白質を発現させることで達成できる。しかし重水は軽水に比べて細胞毒性があり、重水で調製した培地中では菌体の生育・増殖効率が顕著に低下し、目的蛋白質の収量も顕著に低下するのが一般的である。以下、酵母K. lactis発現系で目的蛋白質を高度に重水素化するための培養方法について記載する。
1. 培地・試薬類の調製
- YCB–アセトアミド寒天培地
- NEBの取扱説明書を参照。必要に応じて重水(\(\ce{^{2}H2O}\)、99.8 atom% \(\ce{^{2}H}\))を用いて調製する。
- 1 Mリン酸カリウム緩衝液、10× YNB溶液、500× ビオチン溶液、200× PTM1溶液
- \(\ce{^{2}H2O}\) で、必要な量だけ用時調製する。
- \(\ce{^{15}N}\) 塩化アンモニウム溶液
- 培養前に用時調製。7.5 gの \(\ce{^{15}N}\) 塩化アンモニウム(98 atom% \(\ce{^{15}N}\))を75 mLの \(\ce{^{2}H2O}\) で溶解し、クリーンベンチ内でろ過滅菌。
- D-[\(\ce{^{2}H7}\)]グルコース溶液
- 培養前に必要に応じて用時調製。2 gのD-[\(\ce{^{2}H7}\)]グルコースを50 mLの \(\ce{^{2}H2O}\) で溶解し、クリーンベンチ内でろ過滅菌。
- \(\ce{^{2}H,^{15}N}\) 最小培地
- 500 mLの \(\ce{^{2}H,^{15}N}\) 最小培地を、下記の要領で調製する。2 L容量のファーメンタージャーを空の状態でオートクレーブ滅菌し、恒温乾燥機等に入れてジャー内部の水滴を乾燥させる。クリーンベンチ内にてジャー内に325 mLの \(\ce{^{2}H2O}\)、0.25–0.50 mLの消泡剤Antifoam 204(SIGMA)、50 mLの10× YNB溶液、1 mLの500× ビオチン溶液、50 mLの1 Mリン酸カリウム緩衝液、2.5 mLの200× PTM1溶液、25 mLの \(\ce{^{15}N}\) 塩化アンモニウム溶液(=終濃度5 g/L)、50 mLのD-[\(\ce{^{2}H7}\)]グルコース溶液(=終濃度4 g/L)を無菌的に添加する。
- D-[\(\ce{^{2}H7}\)]グルコース溶液(流加用)
- 培養前に必要に応じて用時調製。6 gのD-[\(\ce{^{2}H7}\)]グルコースを100 mLの \(\ce{^{2}H2O}\) で溶解し、クリーンベンチ内でろ過滅菌。
- \(\ce{^{2}H,^{15}N}\) 最小培地(流加用)
- 1 Lの \(\ce{^{2}H,^{15}N}\) 最小培地を、下記の要領で調製する。1 L容量のメジュームビンを空の状態でオートクレーブ滅菌し、恒温乾燥機内などでボトル内部の水滴を乾燥させる。クリーンベンチ内で650 mLの \(\ce{^{2}H2O}\)、0.5–1.0 mLの消泡剤Antifoam 204(SIGMA)、100 mLの10× YNB溶液、2 mLの500× ビオチン溶液、100 mLの1 Mリン酸カリウム緩衝液、5 mLの200× PTM1溶液、50 mLの \(\ce{^{15}N}\) 塩化アンモニウム溶液(=終濃度5 g/L)、100 mLのD-[\(\ce{^{13}C6}\)]グルコース溶液(=終濃度6 g/L)を、クリーンベンチ内で無菌的に添加する。
- 50% [v/v] エタノール溶液
2. 均一 \(\ce{^{2}H,^{15}N}\) 標識の培養の手順
- K. lactis 形質転換体のシングルコロニーもしくはグリセロールストック少量を滅菌済みの爪楊枝等でピックし、YCB–アセトアミド寒天培地上に画線培養する(30℃で静置しコロニー形成)。この時、\(\ce{^{2}H2O}\) で調製したYCB–アセトアミド寒天培地を用いれば、重水環境に早期に馴化して重水培地中でも比較的増殖や蛋白質発現量が高い株を取得できる可能性がある。
- Freshなシングルコロニーを、5 mLのYPD培地(50 mLコーニングチューブ)に植菌し、30℃でレシプロ振とう培養(250 rpm)、2日間。
- 室温にて3,000 g、5分間の遠心で菌体を遠沈し、2 Lジャー内から \(\ce{^{2}H,^{15}N}\) 培地を数十mLとって、それで菌体を穏やかに再懸濁し、2 Lジャー内の \(\ce{^{2}H,^{15}N}\) 培地に戻す。
- ポアサイズ0.22 μmのシリンジフィルターを通した大気を1.5 L/minで培地中に供給しながら30℃で800 rpmで撹拌培養。この時、ジャー内へ供給する空気から水分を除去するため、ジャーに繋がる空気ホースの途中に、塩化カルシウム顆粒を充填した管を接続しておく。
- ジャーと流加用 \(\ce{^{2}H,^{15}N}\) 最小培地が入ったメジュームビンを、シリコンチューブを介して接続する。そのシリコンチューブにペリスタポンプを接続する(図1)。一定の流速で流加用 \(\ce{^{2}H,^{15}N}\) 最小培地をジャー内の培養液に供給する。この時、メジュームビン内の流加用 \(\ce{^{2}H,^{15}N}\) 最小培地への水分混入を防ぐため、メジュームビンの空気取り込み口に塩化カルシウム顆粒を充填した管を接続しておく。
- 培養終了後、ジャーから培養液を回収し、10,000 gで15分間の遠心により菌体を沈殿させる。目的蛋白質を細胞内に発現させた場合は、遠心上清は廃棄して遠沈した菌体を回収し、菌体を破砕して目的蛋白質の精製へと進む。目的蛋白質を培地中に分泌発現させた場合は、遠心上清(培養後の培地)をポアサイズ0.45 μmのボトルトップフィルターでろ過して、菌体等の夾雑物を除去したうえで培地のみ回収する。筆者らの検討では、この手順により92%以上の \(\ce{^{2}H}\) 標識率を達成できている (3)。次項5)では、さらに酵母形質転換株の重水馴化のための中間培養を1工程挟む培養方法を紹介する。その中間培養ステップを経ることにより、さらに蛋白質発現量が向上し、かつ高効率(95%以上)の \(\ce{^{2}H}\) 標識率を達成できている (4)。もちろんその培養方法は均一 \(\ce{^{2}H,^{15}N}\) 標識にも適用可能である。
5)均一 \(\ce{^{2}H}\) 標識を伴う安定同位体(ILVメチル基選択的 \(\ce{^{1}H,^{13}C}\))標識法
イソロイシン・ロイシン・バリン(ILV)残基のメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識は、それらアミノ酸の前駆体に安定同位体標識を導入したものを培地中に添加して目的蛋白質を発現させることで達成される。大腸菌発現系では適切なα-ケト酸を用いることで問題無く目的の標識が達成されることが知られているが、酵母K. lactisでは、アミノ酸生合成がミトコンドリアに局在する酵素群によって媒介されるため、単純に前駆体を添加しただけでは、前駆体がミトコンドリアに適切に取り込まれない等の障壁により、標識効率が高くならない場合がある。例えばIleの生合成は細胞質にて主に進行するため、Ileのメチル基選択的標識は問題無く達成される。しかしながら、Leu,Valは酵母ミトコンドリア内に局在する酵素群によって生合成されるため、前駆体を添加しただけでは、Leu,Valのメチル基選択的標識はほぼ不可能である。そこで筆者らは、ミトコンドリアに局在するBranched-chain amino acid amino-transferase(BCAT)をK. lactisの細胞質に過剰発現させることにより、Ileの場合と同じくLeu,Valについても充分高い効率でメチル基選択的標識を達成することに成功した(図2)(4)。一方、メチル基が \(\ce{^{1}H,^{13}C}\) 標識されているValを培地に添加すれば、ValがLeuに細胞内で転換され、LVのメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識が可能である (4)。しかしメチル基に選択的に標識が施されたアミノ酸は高価であるため、コストの観点からは、そのようなアミノ酸を使わずにLVのメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識を可能にする工夫は重要である。
目的蛋白質とBCATを共発現する形質転換株の作製の詳細については文献4を参照いただきたいが、前述のように、K. lactisの形質転換株の選別方法は複数の遺伝子を共発現する株の取得効率が高く、このような “共発現” 戦略は目的蛋白質の安定同位体標識だけでなく様々な目的においても有益なK. lactis発現系の特徴の一つである。
1. 培地・試薬類の調製
- YPD培地
- クリーンベンチ内で、YP培地 : 40% [w/v] グルコース溶液の液量比を39 : 1で(グルコースの終濃度が1% [w/v] となるように)混和する(例:YP培地19.5 mLに40% [w/v] グルコース溶液0.5 mL混和。必要量のみを用時調製)。
- YCB–アセトアミド寒天培地
- NEBの取扱説明書を参照。必要に応じて、溶媒は \(\ce{^{2}H2O}\) を用いて調製する。
- 1 Mリン酸カリウム緩衝液、10× YNB溶液、500× ビオチン溶液、200× PTM1溶液
- \(\ce{^{2}H2O}\) で、必要な量だけ用時調製する。
- 塩化アンモニウム溶液
- 培養前に用時調製。2.2 gの塩化アンモニウムを11 mLの \(\ce{^{2}H2O}\) で溶解し、クリーンベンチ内でろ過滅菌。
- D-[\(\ce{^{2}H7}\)]グルコース溶液
- 培養前に必要に応じて用時調製。6 gのD-[\(\ce{^{2}H7}\)]グルコースを60 mLの \(\ce{^{2}H2O}\) で溶解し、クリーンベンチ内でろ過滅菌。
- [methyl-\(\ce{^{13}C}\),3,3-\(\ce{D2}\)]α-ketobutyrate溶液(イソロイシンのδ1メチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識用前駆体)
- 培養前に必要に応じて用時調製。100 mgを100 mLの \(\ce{^{2}H2O}\) で溶解し、クリーンベンチ内でろ過滅菌。
- [3-methyl-\(\ce{^{13}C}\), 3,4,4,4-\(\ce{D4}\)]α-ketoisovalerate溶液(バリン・ロイシンのメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識用前駆体)
- 培養前に必要に応じて用時調製。100 mgを100 mLの \(\ce{^{2}H2O}\) で溶解し、クリーンベンチ内でろ過滅菌。
- \(\ce{^{2}H}\) 最小培地(馴化培養用)
- 100 mLの \(\ce{^{2}H}\) 最小培地を、下記の要領で調製する。500 mL容量のフラスコを用意し、その口にシリコン栓をして(そのシリコン栓には2つの穴を開け、ステンレス製のニードルを2本通しておく。一方のニードルはフラスコの底にギリギリ接しないくらいの長さとし、培地中に空気を供給するためのニードルとして使う。もう一方のニードルは排気用のため短めで(シリコン栓を貫通していれば)よい)、空の状態でオートクレーブ滅菌し、恒温乾燥機等に入れてフラスコ内部の水滴を乾燥させる。クリーンベンチ内にてそのフラスコ内に68 mLの \(\ce{^{2}H2O}\)、0.05 mLの消泡剤Antifoam 204(SIGMA)、10 mLの10× YNB溶液、0.2 mLの500× ビオチン溶液、10 mLの1 Mリン酸カリウム緩衝液、0.5 mLの200× PTM1溶液、1 mLの塩化アンモニウム溶液(=終濃度2 g/L)、10 mLのD-[\(\ce{^{2}H7}\)]グルコース溶液(=終濃度10 g/L)を無菌的に添加する。
- \(\ce{^{2}H}\) 最小培地(本培養用)
- 400 mLの \(\ce{^{2}H}\) 最小培地を、下記の要領で調製する。2 L容量のファーメンタージャーを空の状態でオートクレーブ滅菌し、恒温乾燥機等に入れてジャー内部の水滴を乾燥させる。クリーンベンチ内にてジャー内に0.2–0.4 mLの消泡剤Antifoam 204(SIGMA)、40 mLの10× YNB溶液、0.8 mLの500× ビオチン溶液、40 mLの1 Mリン酸カリウム緩衝液、2 mLの200× PTM1溶液、4 mLの塩化アンモニウム溶液(=終濃度2 g/L)、24 mLのD-[\(\ce{^{2}H7}\)]グルコース溶液(=終濃度6 g/L)を無菌的に添加する。さらに、イソロイシンのδ1メチル基を選択的に \(\ce{^{1}H,^{13}C}\) 標識する場合は40 mLの[methyl-\(\ce{^{13}C}\),3,3-\(\ce{D2}\)]α-ketobutyrate溶液(=終濃度100 mg/L)、バリン・ロイシンのメチル基を選択的に \(\ce{^{1}H,^{13}C}\) 標識する場合は40 mLの[3-methyl-\(\ce{^{13}C}\), 3,4,4,4-\(\ce{D4}\)]α-ketoisovalerate溶液(=終濃度100 mg/L)を無菌的に添加する。\(\ce{^{2}H2O}\) を加えて全量400 mLにする。
- \(\ce{^{2}H}\) 最小培地(流加用)
- 600 mLの \(\ce{^{2}H}\) 最小培地を、下記の要領で調製する。1 L容量のメジュームビンを空の状態でオートクレーブ滅菌し、恒温乾燥機内などでボトル内部の水滴を乾燥させる。クリーンベンチ内で0.3–0.6 mLの消泡剤Antifoam 204(SIGMA)、60 mLの10× YNB溶液、1.2 mLの500× ビオチン溶液、60 mLの1 Mリン酸カリウム緩衝液、3 mLの200× PTM1溶液、6 mLの \(\ce{^{15}N}\) 塩化アンモニウム溶液(=終濃度2 g/L)、26 mLのD-[\(\ce{^{2}H7}\)]グルコース溶液(=終濃度4 g/L)を無菌的に添加する。さらに、イソロイシンのδ1メチル基を選択的に \(\ce{^{1}H,^{13}C}\) 標識する場合は60 mLの[methyl-\(\ce{^{13}C}\),3,3-\(\ce{D2}\)]α-ketobutyrate溶液(=終濃度100 mg/L)、バリン・ロイシンのメチル基を選択的に \(\ce{^{1}H,^{13}C}\) 標識する場合は60 mLの[3-methyl-\(\ce{^{13}C}\), 3,4,4,4-\(\ce{D4}\)]α-ketoisovalerate溶液(=終濃度100 mg/L)を無菌的に添加する。\(\ce{^{2}H2O}\) を加えて全量600 mLにする。
- 50% [v/v] エタノール溶液
2. 均一 \(\ce{^{2}H}\) 標識・メチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識の培養の手順
- K. lactis形質転換体(目的蛋白質とBCATの共発現株)のシングルコロニーもしくはグリセロールストック少量を滅菌済みの爪楊枝等でピックし、YCB–アセトアミド寒天培地に画線培養する(30℃)。この時、\(\ce{^{2}H2O}\) で調製した YCB–アセトアミド寒天培地を用いれば、重水環境に早期から馴化して重水培地中でも比較的増殖が良い株を取得できる可能性がある。
- Freshなシングルコロニーを、10 mLのYPD培地(50 mLコーニングチューブ)に植菌し、30℃でレシプロ振とう培養(250 rpm)、3日間。
- 室温にて3,000 g、5分間の遠心で菌体を遠沈し、500 mL容量フラスコから \(\ce{^{2}H}\) 培地(馴化培養用)を適当量とって、それで菌体を穏やかに再懸濁し、500 mLバッフル付フラスコの \(\ce{^{2}H}\) 培地に戻す。そのフラスコの口を、ニードルを通したシリコン栓(前項を参照)で栓をする。
- 500 mL容量フラスコのシリコン栓に挿したニードル2本のうち、先端が培地中に浸っているニードルに空気ポンプのホースを接続し、ポアサイズ0.22 μmのシリンジフィルターを通した大気を0.5 L/minで培地中に供給しながら、30℃で250 rpmで撹拌培養(3日間)。この時、培地中へ供給する空気から水分を除去するため、空気ポンプとフラスコを繋ぐ空気ホースの途中に、塩化カルシウム顆粒を充填した管を接続しておく。
- 室温にて3,000 g、5分間の遠心で菌体を遠沈し、ジャーから \(\ce{^{2}H}\) 培地を適当量とって、それで菌体を穏やかに再懸濁し、ジャー内の \(\ce{^{2}H}\) 培地に加える。
- ジャーと流加用 \(\ce{^{2}H}\) 最小培地が入ったメジュームビンを、シリコンチューブを介して接続する。そのシリコンチューブの途中にペリスタポンプを接続する(図1)。ポアサイズ0.22 μmのシリンジフィルターを通した大気を1.5 L/minで培地中に供給しながら、30℃で800 rpmで撹拌培養(3日間)。この間、一定の流速で流加用 \(\ce{^{2}H}\) 最小培地(600 mL)をジャー内の培養液に供給する。例えば、流速8 mL/hで添加すれば75時間で600 mL全量をジャーに供給することとなる。培養期間を考慮して流速を決定する。流加用 \(\ce{^{2}H}\) 最小培地が入ったメジュームビン内へ大気中の水分が混入することを防ぐため、メジュームビンの空気取り込み口に塩化カルシウム顆粒を充填した管を接続しておく。
- 培養終了後、ジャーから培養液を回収し、10,000 gで15分間の遠心により菌体を沈殿させる。目的蛋白質を細胞内に発現させた場合は、遠心上清は廃棄して遠沈した菌体を回収し、菌体を破砕して目的蛋白質の精製へと進む。目的蛋白質を培地中に分泌発現させた場合は、遠心上清(培養後の培地)をポアサイズ0.45 μmのボトルトップフィルターでろ過して、菌体等の夾雑物を除去したうえで培地のみ回収する。
最後に
K. lactisだけでなく、かねてから広く利用されているメタノール資化性酵母P. pastorisを宿主とする発現系も洗練されたキットとして市販されており、初心者でも酵母発現系を導入しやすくなっている。また、NMR測定試料のための安定同位体標識のバリエーションやコストも大腸菌発現系のそれと遜色ないところまで発展してきた。高難度蛋白質の大量発現と安定同位体標識において、もはや酵母発現系は大腸菌発現系と並んで第一選択の手法である。本稿が、高難度蛋白質の大量発現もしくは安定同位体標識において大腸菌発現系の “壁” を感じておられる研究者の方々の一助となれば幸いである。
工夫とコツ
P. pastoris発現系における恒常的な目的蛋白質の発現誘導
P. pastoris発現系は、一般的な方法(pPICベクターを使用しAOXプロモーターの制御下で目的蛋白質の遺伝子を発現誘導)では、目的蛋白質の発現誘導にはメタノールの添加が必要であるが、glyceraldehyde-3-phosphate dehydrogenase(GAP)プロモーターの制御下で目的蛋白質を発現誘導するpGAPベクターを用いれば、P. pastorisでもK.lactis発現系のようにグルコース含有培地で恒常的な目的蛋白質の発現誘導が可能である (5,6)。
共発現株の作製
複数の異なる蛋白質をK. lactis内で共発現させる場合、形質転換の方法自体は取扱説明書に記載されている定法に従い、複数種類の目的遺伝子を同時に形質転換するだけでよい。ある一定の確率で共発現株が得られるが、筆者らの経験では形質転換コロニーの5%以上は共発現株となる。K. lactisのゲノムに複数の目的遺伝子が挿入されていることはcolony-direct PCR法により検証できる。その手順の詳細はNEBの取扱説明書を参照いただきたい。
リン酸–金属錯体の白色沈殿の発生
PTM1溶液とリン酸緩衝液を混和すると、PTM1溶液の金属とリン酸が不溶性の錯体を形成し、白い沈殿が発生する。これは特にpHが高いと生じやすい。この沈殿は菌体に傷害を与えることはなく、菌体の生育・増殖が顕著に低下することはないが、流加用の培地中に沈殿が大量に発生するとシリコンチューブ内が詰まり、培地の安定供給が困難となる。その場合は、リン酸緩衝液もしくはPTM1溶液の添加量を減らす等の対策を講じる。また、目的蛋白質を \(\ce{^{2}H}\) もしくは \(\ce{^{13}C}\) で標識しない場合は、リン酸緩衝液を他の緩衝液(トリス等)に変更してもよい。
培養中の培地のpHと溶存酸素濃度
培養中に培地のpHおよび溶存酸素濃度をセンサーでモニターし、pHおよび溶存酸素濃度を設定値に維持することが可能であれば適用したほうが好ましい。ちなみに、培地中から炭素源が枯渇すると、溶存酸素濃度の値が急激に上下動する(\(\ce{O2}\) スパイク)現象が観察される。Fed-batch培養では、\(\ce{O2}\) スパイクが発生しないぎりぎりのタイミングでfreshな培地を供給することを継続的に達成できるように、流加する培地の組成や添加速度を調整するのが、最小限の炭素源の消費量で最大限のバイオマスを得る最適な培養条件である。そのうえでも溶存酸素濃度のモニターは培養条件の最適化において有用である。
ジャーファーメンターもしくはカルチャーバッグの重要性
酵母の十分な増殖と蛋白質発現には、炭素源(糖)および窒素源の量とともに、培地の溶存酸素濃度が最もクリティカルに影響する。そのため、特に安定同位体標識のための最小培地のように、貧栄養性で菌体の増殖には厳しい環境(重水培地の場合はさらに細胞毒性があり生育には厳しい条件となる)での培養や、コスト上の制約から炭素源の添加量を出来る限り抑えたい場合には、培養中に十分量の酸素を培地に供給することが極めて重要となる。したがって、酵母発現系を用いた安定同位体標識蛋白質の発現のための培養では、大腸菌において用いられるバッフル付きフラスコではなく、ジャーファーメンターもしくはカルチャーバッグの使用がほぼ不可欠である。それらの装置を使用できない場合でも、\(\ce{^{2}H}\) 標識の際の中間培養(前述)のように、バッフル付フラスコを用いた培養中に空気ポンプで大気を培地に供給することで、ジャーファーメンターもしくはカルチャーバッグに準ずる培養環境を整えることは可能である (7)。
融合蛋白質の利用
K. lactis発現系はマルトース結合蛋白質(Maltose-binding protein, MBP)を大量に発現することができ、K. lactis発現系構築のポジティブコントロールとして有用である。MBP遺伝子をコードしたpKLACプラスミド(pKLAC1-malE)がNEBのK. lactis Protein Expression Kitに含まれている。
MBPは大腸菌発現系でも目的蛋白質の “SET”(solubility enhancement tag)として用いられ、目的蛋白質の発現量および可溶性を向上させることが知られている。また、MBPはアミロース樹脂により簡単かつ高純度にワンステップ精製が可能であるため、目的蛋白質に融合すればその精製タグとしても有用である。
またMBPは、分泌シグナル配列を付加することにより、大腸菌において細胞質だけでなくペリプラズム画分へも発現させることができ、酵母においても細胞質発現もしくは分泌発現のどちらも可能である。
MBPは43 kDaという溶液NMRの測定対象としては高分子量の部類に入るが、線幅がシャープで分散も良い高質な溶液NMRスペクトルが得られるため、安定同位体標識法の開発におけるモデル蛋白質としても有用である。
筆者らは、pKLAC1-malEベクター内のMBP遺伝子の下流に目的蛋白質のcDNAを挿入したコンストラクトを作製(pKLAC1-malEベクターのNot I/Stu Iサイト間に目的cDNAを挿入したプラスミドをまず作製し、その後MBP遺伝子と目的cDNA間に残っているストップコドンをMutagenesis PCR法でdeletionする)し、目的蛋白質のN末端にMBPを融合して発現させることを行っている。
MBP以外にも、酵母発現系でヒト血清アルブミン(Human Serum Albumin; HSA)、SUMO、GST等が目的蛋白質のSETとして利用された例がある。目的蛋白質単独での発現量が十分でなかった場合、SETの融合発現を検討してみることをお勧めする。
プロテアーゼ欠損酵母株の利用
酵母の液胞に存在するプロテアーゼはその活性が強力で、目的蛋白質を細胞内で発現させる場合や酵母細胞を破砕して目的蛋白質を抽出・精製する場合には、その過程で目的蛋白質の分解が生じるリスクが高まる。精製中の分解を抑制するには、菌体破砕のためのバッファー類に市販のプロテアーゼインヒビターカクテルを添加しておくことは必須である。それに加えて、特定のプロテアーゼを欠損したK. lactis株がNEBから市販されている。
文献
- Sugiki, T., Shimada, I., and Takahashi, H. J. Biomol. NMR, 42, 159–162 (2008)
- Takahashi, H. & Shimada, I. J. Biomol. NMR, 46, 3–10 (2010)
- Sugiki, T., et al. Methods Mol. Biol., 831, 19–36 (2012)
- Miyazawa-Onami, M., et al. J. Biomol. NMR, 57, 297–304 (2013)
- Zhang, A-L., et al. Mol. Biol. Rep., 36, 1611–9 (2009)
- 櫻井一正, 蛋白質科学会アーカイブ, 1, e018 (2008)
- 村木三智郎, 蛋白質科学会アーカイブ, 7, e078 (2014)
-
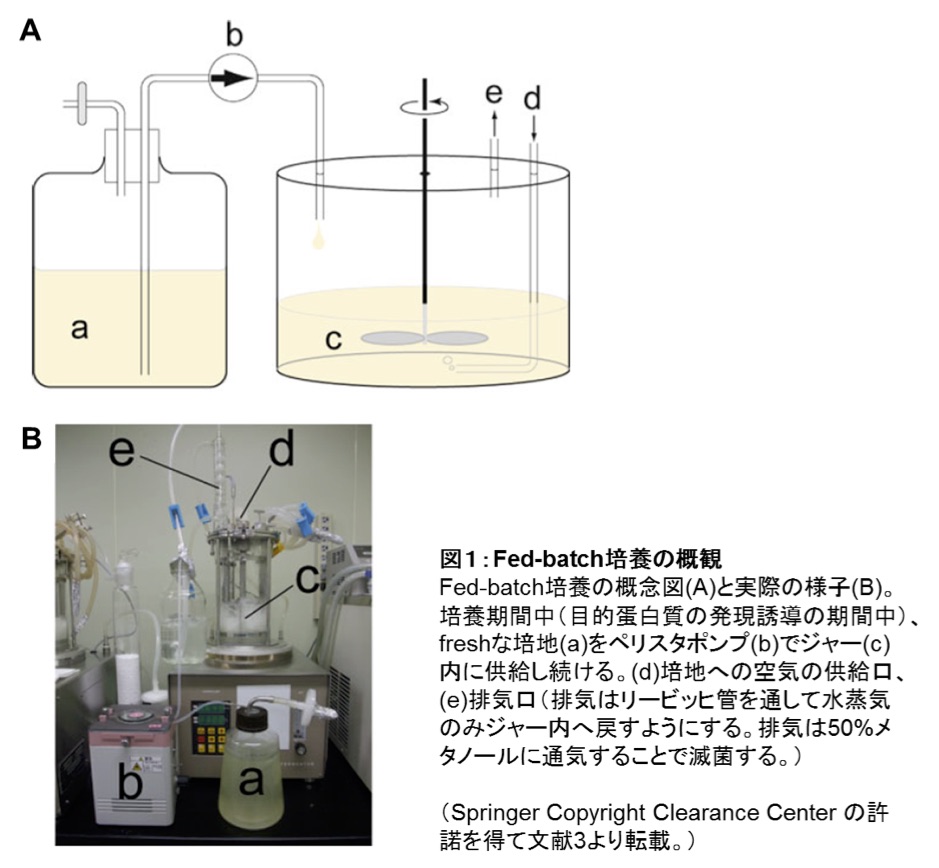
図1:Fed-batch培養の概観
Fed-batch培養の概念図(A)と実際の様子(B)。培養期間中(目的蛋白質の発現誘導の期間中)、freshな培地(a)をペリスタポンプ(b)でジャー(c)内に供給し続ける。(d)培地への空気の供給口、(e)排気口(排気はリービッヒ管を通して水蒸気のみジャー内へ戻すようにする。排気は50%メタノールに通気することで滅菌する。)
(Springer Copyright Clearance Centerの許諾を得て文献3より転載。) -
![MBPの均一\(\ce{^{2}H}\)・ILVメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識酵母K. lactis発現系により調製した均一 \(\ce{^{2}H}\)・ILVメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識MBPの \(\ce{^{1}H}\)-\(\ce{^{13}C}\) HMQC NMRスペクトル。Ile(δ1)およびLeu・Valのメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識は、それぞれのアミノ酸の前駆体である[methyl-\(\ce{^{13}C}\), 3,3-\(\ce{D2}\)]α-ketobutyrateおよび[3-methyl-\(\ce{^{13}C}\), 3,4,4,4-\(\ce{D4}\)-ketoisovalerateを蛋白質発現誘導中に添加することで行った。大腸菌発現系ではILVともに高効率で標識される(A)が、K. lactis発現系ではIleは高効率で標識できるもののLeu,Valはほとんど標識されない(B)。しかし前駆体物質をLeu,Valに変換するBCAT酵素であるilvEもしくはBat2を目的蛋白質と共発現させることにより、K. lactis発現系でのLeu,Valの標識が可能(C,D)。図中右下の “x5” は、スペクトルの切り出しの深さが、無表示のスペクトルに比べて5倍深い位置であることを示す。(Springer Copyright Clearance Centerの許諾を得て文献4より転載。)](/archives/images/Kluyveromyces_01/Kluyveromyces_01_Fig_02.png)
図2:MBPの均一\(\ce{^{2}H}\)・ILVメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識
酵母K. lactis発現系により調製した均一 \(\ce{^{2}H}\)・ILVメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識MBPの \(\ce{^{1}H}\)-\(\ce{^{13}C}\) HMQC NMRスペクトル。Ile(δ1)およびLeu・Valのメチル基選択的 \(\ce{^{1}H,^{13}C}\) 標識は、それぞれのアミノ酸の前駆体である[methyl-\(\ce{^{13}C}\), 3,3-\(\ce{D2}\)]α-ketobutyrateおよび[3-methyl-\(\ce{^{13}C}\), 3,4,4,4-\(\ce{D4}\)-ketoisovalerateを蛋白質発現誘導中に添加することで行った。大腸菌発現系ではILVともに高効率で標識される(A)が、K. lactis発現系ではIleは高効率で標識できるもののLeu,Valはほとんど標識されない(B)。しかし前駆体物質をLeu,Valに変換するBCAT酵素であるilvEもしくはBat2を目的蛋白質と共発現させることにより、K. lactis発現系でのLeu,Valの標識が可能(C,D)。図中右下の “x5” は、スペクトルの切り出しの深さが、無表示のスペクトルに比べて5倍深い位置であることを示す。
(Springer Copyright Clearance Centerの許諾を得て文献4より転載。)
