

概要
1995年にインフルエンザ菌で全ゲノムの配列が決定されて以来、次々と生物種のゲノム配列が決定されている。興味深いことに全遺伝子に対する膜蛋白質の数の占める割合は、大腸菌からヒトに至るまでおおむね30%にも及ぶ。膜蛋白質は水にほとんど溶けない生体リン脂質膜に埋め込まれており、物理化学的性質が水溶性蛋白質と大きく異なる。そのために水溶性蛋白質で培われてきた知見が適用できないことが多く、水溶性蛋白質ほど理解が深まってはいない。個々の膜蛋白質を「蛋白質科学」するためには生体膜に含まれる膜蛋白質、リン脂質を界面活性剤で可溶化し、分離・精製しなければならない。また、膜蛋白質の機能解析を行う際には、リン脂質二重膜に再度埋め込むことが必要な場合もある。
本プロトコールは#040「大腸菌を用いた組換え膜タンパク質の可溶化、精製、リポソームへの再構成」中村寛夫, 蛋白質科学会アーカイブ, 1, e040 (2008) を改訂し、新たにMembrane scaffold protein(MSP)を用いた脂質ナノディスクへの再構成法を加えた増補版である(概略は図1)。著者は脂質や再構成系の専門家ではない。いち利用者として、ヘムを感知する病原菌のセンサーキナーゼChrS (1,2) での研究例を紹介することで、新たに膜蛋白質に取り組む研究者が再構成系を利用した蛋白質科学の研究に「まずはやってみよう」と踏み出すための一助を供したい。
装置・器具・試薬
- 大腸菌BL21(DE3)/pLysS
- ベクターpRSET-A
- pMSP1E3D1(Addgene社)
- イソプロピル-β-チオガラクトピラノシド(IPTG)
- 高速遠心機(各社)
- 超遠心機(各社)
- フレンチプレス(大岳製作所)
- Ni-NTA樹脂(QIAGEN社)10 mL
- チップ式超音波細胞破砕機(各社、出力200 W)
- 界面活性剤
- オクチルグルコシド(n-octyl-β-glucoside)(OG)
- シュクロースモノラウレート(sucrose monolaurate)(SM)
- ドデシルマルトシド(n-dodecyl-β-D-maltoside)(DDM)
- デシルマルトシド(n-decyl-β-D-maltoside)(DM)
- トライトンX-100(Triton X-100)(TX-100)
- コール酸ナトリウム
- プレシジョンプロテアーゼ(GE Healthcare社)
- 大腸菌リン脂質(Avanti Polar Lipids社)
- Bio-Beads SM-2(BioRad社)
実験手順
- 膜蛋白質調製編
- 組換え膜蛋白質を含む大腸菌細胞膜の調製(0.5日)
- 組換え膜蛋白質の可溶化、精製(3–4日)
- プロテオリポソーム編
- リポソームの調製(2日)
- リポソームへの再構成(2日)
- ナノディスク編
- MSP1E3D1の精製(培養を含めて4日)
- ナノディスクへの再構成(1–2日)
実験の詳細
膜蛋白質調製編
組換え膜蛋白質を含む大腸菌細胞膜の調製
第1日
−80℃で保存した菌体10 gに対して200 mLの菌体破砕バッファー(30 mM Tris HCl pH8、20% (w/v) sucrose、以下pHは室温で調整。%はすべてw/v)を加えて、ペレットを完全に分散させる。ここに1 mLの20 mg/mL lysozyme、2 mLの0.5 M EDTA-Na pH8を添加し、4℃で15分間攪拌する。続いて15,000 rpm(34,000 x g)で15分間遠心し、上清を50 mL捨てる。残ったバッファーと沈殿した菌体を取り出し、120 MPa(1,200 kg/cm2程度)の圧力でFrench Pressを2回行う。破砕した菌体溶液と等量の3 mM EDTAを加えた後に1/300量の300 mM PMSF(phenylmethylsulfonyl fluoride;52 mg/mL DMSO)を添加する。12,000 rpm(22,000 x g)で15分間遠心する。上清を回収して超遠心用のチューブに移し、ベックマンアングルローター45Tiを用いて40,000 rpm(200,000 x g)、4℃で1時間超遠心する。超遠心の上清を捨てた後、ガラス棒でペレットを回収し蛋白質濃度がおよそ20–30 mg/mLになるように細胞膜保存バッファー(25 mM Tris HCl pH7.5、10%sucrose)を加える。氷冷しながらホモジナイズし、溶液を−80℃で保存する。菌体10 gから150–200 mgの全膜蛋白質が得られる。
組換え膜蛋白質の可溶化、精製
なんといっても、対象となる組換え膜蛋白質が大腸菌細胞膜で機能をもった状態で発現されていることが望ましい。本研究ではChrSがヘム依存的なリン酸化活性を保持していることを確認済みである。次に、可溶化の条件検討が必要である。マイルドな非イオン性界面活性剤4種類(OG、DM、DDM、SM)で可溶化を試みた。
膜蛋白質画分の終濃度が2 mg/mLとなるように 2×可溶化バッファー(50 mM Tris-HCl pH7.5、10%glycerol、300 mM NaCl)、蒸留水、界面活性剤(終濃度が1.2%)を加えた(可溶化の確認であれば、全量はおよそ1 mLで十分である)。4℃で1時間攪拌した溶液を200,000 x g、4℃で1時間超遠心し、上清を回収した。そして可溶化された蛋白質を SDS-12.5%PAGEによって確認した。図2よりOGではChrS蛋白質は可溶化できないことがわかる。本研究では、再構成の際に透析で取り除きやすいDM(臨界ミセル濃度0.08%)を用いることにした。
第1日
200 mgの全膜蛋白質を1.2%DMで可溶化して得た上清100 mLをバッファーA(25 mM Tris-HCl pH7.5、5%glycerol、150 mM NaCl、0.2%DM)で平衡化したNi-NTAカラム(樹脂容量10 mL)に添加する。バッファーの流速が1 mL/minとなるように調節し、100 mLのバッファーAで洗浄する。続いてカラムにバッファーA 80 mLとバッファーB(バッファーAにイミダゾール400 mMを含む)80 mLでグラジエントをかけながら添加してChrS-PreScission-10×Hisを溶出する(図3)。予め可溶化上清とバッファーAにイミダゾールを50 mMになるように加えておき50–400 mMのグラジェントによる溶出でもよい結果が得られる(工夫とコツ、発現系について参照)。
第2日
得られた蛋白質画分に関してSDS-PAGEを行い、ChrS-PreScission-10xHisに相当するバンドが確認された画分を回収する(10–15 mg)(図4;レーン2)。濃縮器セントリプレップYM-50(Millipore社)を用いて、蛋白質濃度が1–2 mg/mL程度になるまで遠心機himacCF16RX、スウィングローターT5SSを使い2,500 rpm(1,100 x g)4℃で濃縮し、ChrSの1/100重量のPreScission protease(1 unit/μg)を加える。この溶液を透析チューブに入れ1 Lの透析バッファー(50 mM Tris-HCl pH7.5、5%glycerol、150 mM NaCl、0.2%DM)中で4℃、3時間透析を行う。もうひとつの透析バッファー1 Lに移し一晩透析をする(ヒスタグ切断と透析によるイミダゾール除去を同時に行っている)。
第3日
プロテアーゼ処理後の透析チューブの中身の一部(2–4 μg相当)をSDS-PAGEにかけ切断を確認した後(図4;レーン3)、その溶液をNi-NTA カラム(樹脂容量10 mL)に直接添加した。素通り画分とバッファーA 10 mLでの洗浄画分を回収し、さらにバッファーA 50 mLとバッファーB 50 mLでグラジエントをかけながら溶出された画分も回収した(ヒスタグのないChrSもNi-NTA樹脂に弱く結合するようである)。各画分をSDS-PAGEにかけ、ヒスタグ切断が確認されたChrSの画分を回収する。蛋白質濃度がおよそ2 mg/mLになるまでセントリプレップYM-30を用いて2,500 rpm(1,100 x g)4℃で濃縮する。最終的な蛋白質濃度を測定して−80℃で保存する。濃縮が終わらなかったら翌日に続ける。ヒスタグを切断した最終精製標品を図4のレーン4に示す。
プロテオリポソーム編
膜蛋白質をリン脂質膜小胞に埋め込み、得られたプロテオリポソームで物質やイオン輸送の機能解析を行った研究論文は1970年代中頃から見られる。本稿では多くの膜蛋白質で活性を回復させた実績のある界面活性剤を用いたリポソームへの再構成法を紹介する (3)。別法として、膜蛋白質、脂質を有機溶媒で抽出し、混合後、有機溶媒を除くことで脂質に再構成する方法もあるが、膜蛋白質の変性、失活を伴うことが多い。また、界面活性剤を加えずに凍結・融解、超音波処理を行う方法もあるが、精製した膜蛋白質溶液に持ち込みの界面活性剤が含まれているので原理的には界面活性剤を介していると解釈できる。
リポソームの調製
第1日
クロロホルムに溶解しているAvanti社大腸菌リン脂質40 mg分を遠心エバポレーターで減圧濃縮しクロロホルムを除去する。1.5%OGを含む50 mM Tris-HCl pH7.5溶液5 mLを加え、ボルテックスで溶解させる。溶けない場合はOGを3%にする。次に、透析バッファー(50 mM Tris-HCl pH7.5)1 L中で2時間の透析を2回行う。3回目は4℃で一晩透析し、界面活性剤を取り除くことでリポソームを調製する。
第2日
リポソーム試料液の容積を測り、リン脂質濃度を調べる(6–8 mg/mLとなる)(正確な濃度決定にはリンの定量が必要である。工夫とコツ、リポソームの調製について参照。)エッペンドルフチューブに分注して液体窒素で凍らせ、室温でゆっくり溶かすという作業を2回繰り返し、最後に再度液体窒素で凍らせて−80℃に保存する。
リポソームへの再構成
第1日
リポソームを使用する時には室温でゆっくり溶かして用いる。この状態ではリポソーム径が大きいか、または凝集体になっている。用途によって、窒素気相で超音波処理(チップ式、バス式、共に可。短時間ならば室温でもよい)やMini-Extruder(Avanti Polar Lipids社)によるフィルターろ過で粒子径を小さく、均一化しておく。
膜蛋白質とリン脂質は重量比1 : 20–100で混合する(輸送活性測定など1分子埋め込みで十分に閉じた小胞を必要とする場合は蛋白質に対して重量比で脂質を100–200倍加える)。
溶液組成
| ChrS(2.4 mg/mL) | 0.05 mL |
| 大腸菌リポーム(8mg/mL) | 0.3 mL |
| 再構成透析バッファー(下記参照) | 0.115 mL |
| 10%OG | 0.03 mL |
| 1M DTT(dithiothreitol) | 0.005 mL |
| total | 0.5 mL |
上記の溶液を調製後15分間室温で放置し、透析チューブに移す。その後500 mLの再構成透析バッファー(50 mM Tris-HCl pH 7.5、50 mM KCl、5%glycerol)を2つ用意し、4℃で2回(1回目は2–3時間、2回目は一晩)透析する。(工夫とコツ、リポソームへの再構成について参照)
第2日
再構成したプロテオリポソームを回収し、分注して−80℃に保存する。
精製したChrSは界面活性剤存在下では自己リン酸化活性を失っているが、このようにしてリポソームに再構成したChrSはヘムに依存した高い自己リン酸化活性を回復していることがわかる(図5)。
脂質ナノディスク(Nanodiscs)編
脂質はほとんど水に溶けないため溶液中では二重膜層になる。リポソームは内腔をもつ小胞なので生体膜を模倣した膜蛋白質研究(例えば、物質輸送反応の解析)を可能とする。しかし、小胞の径が数百nm以上あり光散乱、沈降などが生じるため、物理化学的測定が困難な場合もある。これに対して、米国イリノイ大のSligar博士らが開発したナノディスク(Nanodiscs)はヒト由来リポプロテインAを改変したMembrane scaffold protein(MSP)とリン脂質からなる円盤状粒子であり、さまざまな遺伝子改変により9–15 nm径のものが得られている(本研究で用いるMSP1E3D1は12 nmの円盤が形成される)(4)。ナノディスクへの膜蛋白質埋め込みは水溶性状態で膜蛋白質の脂質環境を維持できる新しい再構成法であり、可視吸収やNMRなど各種分光測定に利用されている。MSPの発現プラスミドは非営利研究機関であれば送料を含め1万円ほどでAddgene社から入手できる。
MSP1E3D1の精製
第1日
20 mLのLB(1 LあたりBactotrypton 10 g、yeast extract 5 g、NaCl 5 gに0.5%グルコース、50 μg/mLカナマイシンを含む)にpMSP1E3D1を移入したBL21(DE3)のコロニーひとつを植菌する。37℃で4–6時間培養し(OD600 nm,0.6–0.8)、4℃で一晩保存する。
第2日
2 LのTB(1 LあたりBactotrypton 12 g、yeast extract 24 g、\(\ce{KH2PO4}\) 2.3 g、\(\ce{K2HPO4}\) 12.5 g、グリセロール4 gに 20 μg/mLカナマイシンを含む;リン酸塩以下は個別に滅菌する)に前培養液を移し、37℃で3–4時間培養する(Shimazdu UV2500,OD600 nm,0.6–0.8;ただし、2くらいになっても問題はない。同じ培養液でも使用する分光器によって異なる濁度を示すおそれがあるので注意されたし)。1 mMになるようにIPTGを加え、1時間後、28℃にして3.5–4時間培養を続ける。遠心機で集菌し−80℃で保存する。湿重量で20 gくらい得られる。
第3日
菌体10 gを20 mMリン酸ナトリウムバッファー(pH7.4、1 mM PMSF)90 mLに溶融し、10%(w/v) TX-100を1%になるように加える。撹拌後、氷冷しながらチップ式超音波処理を出力200 W、10秒、6回で行う。30,000 x g 30分の遠心で得られた上清を低温室に設置したNi-NTAカラム(樹脂容量30 mL)に添加する。バッファーC(40 mM Tris-HCl pH8、300 mM NaCl)に1%になるようにTX-100を加えたバッファーC-TX 120 mLでカラムを洗浄する。以下のコール酸、イミダゾールを含むバファーの最終pHはHClやNaOHで8に調整しておく。次いで、同量のバッファーC-Ch(50 mMコール酸ナトリウムを含むバッファーC)、バッファーC、バッファーC-Im50(50 mMイミダゾールを含む)で順次洗浄する。100 mLのバッファーC-Im300(300 mMイミダゾールを含むバッファーC)で溶出し、MSPを含む画分を回収する。2 Lの10 mM Tris-HCl pH7.6、100 mM NaCl、1 mM EDTA-Naで二時間ずつ2回透析する。3回目の透析は一晩行う。
第4日
得られたMSP1E3D1の濃度をAbs280 nm, 0.9/mg/mL(文献4のε280,29.9 mM−1cm−1から換算)を基に決定した後、−20℃か−80℃で保存する。100 mgほどの精製標品が得られる。工夫とコツ、MSPについて参照
ナノディスクへの再構成
ナノディスクには2分子のMSPが含まれるので、膜蛋白質が過不足なく取り込まれるように0.6–0.7等量加える。ChrSは二量体が機能単位なのでモル比MSP : ChrS = 1 : 1.2–1.5で加える。また、MSP対リン脂質はモル比1 : 120(重量比1 : 3–5くらい)で加える(本稿では1 mg/mL = 1.3 mM換算)。ナノディスクを生成するためには、界面活性剤で完全に脂質を可溶化して、MSP、膜蛋白質との複合ミセルを形成させてから界面活性剤を除去しなければならない。なお、過剰な膜蛋白質と脂質はプロテオリポソームや凝集体になるため、これらは超遠心やゲルろ過カラムで分離するとよい。
第1日目
溶液組成
| MSP1E3D1(50 μM,1.7 mg/mL) | 0.1 mL |
| ChrS(40 μM,1.8 mg/mL) | 0.188 mL |
| 大腸菌リポソーム(10 mM,8 mg/mL) | 0.06 mL |
| 1 M TrisHCl,pH7.5 | 0.025 mL |
| 1 M KCl | 0.025 mL |
| 10%DM | 0.005 mL |
| 1 M DTT | 0.005 mL |
| \(\ce{H2O}\) | 0.092 mL |
| total | 0.5 mL |
上記の溶液を調製後、室温で1時間ゆっくり回転撹拌してから、メタノール・蒸留水で洗浄したBio-Beads SM-2をおよそ0.3 mL(2/3容量)を秤り取ったエッペンドルフ型チューブに移す。さらに1時間ゆっくり回転撹拌する。エッペンチューブの底付近(肉薄の部分)に26G以下の注射針で穴をあける(くれぐれも怪我をしないように!図6)。フタに11 mm径の穴をあけた17 mL遠心チューブに差し入れてから(図7)、エッペンチューブのフタにも穴をあける。スウィングローターを用い180 x g(1,000 rpm)で1分間遠心して濾過液を回収し、再度Bio-Beads SM-2を含むエッペンチューブに移す。同じ操作をして、界面活性剤を除去した濾過液を回収する。必要ならば透析によりバッファー組成を変える。この透析は徹底的に界面活性剤を除去する効果もある。最後に超遠心(200,000 x g、30分)による上清の回収やゲルろ過カラム(Superdex 200HR 10/30やTSK G5000 PWXL)による水溶性ナノディスク画分の回収を行う。工夫とコツ、再構成したナノディスクについてを参照。
工夫とコツ
発現系について
本プロトコールではベクターpRSET-Aの多コピー性、T7プロモーターのみを利用する。つまり、もともとベクターに付与されているヒスタグやプロテアーゼ切断部位は取り除き、ChrSのC末端にin-frameでPreScission-10×Hisを連結したDNAをNdeI、HindIII部位に挿入した(図8)。
ChrSはN末に6回膜貫通領域をもち、C末が水溶性ヒスチジンキナーゼドメインである。そこで、細胞膜への局在を損ねないように、ヒスタグはC末に融合させた。C末にタグを付加する場合には、PreScissionプロテアーゼで切断後も切断認識配列の一部Leu-Phe-Glnが残ってしまうのが欠点ではある。しかし、ChrSではヘム受容や自己リン酸化の機能は損なわれないようである。
大腸菌膜蛋白質にはNi-NTA樹脂に吸着し、100mM程度のイミダゾールで溶出される多くの蛋白質群がある(しかも、よりによってヘム蛋白質も含まれている)。そこで組換え蛋白質のNi-NTA樹脂への保持力を高めるために、通常の6xHisではなく10×Hisタグを付加し高濃度イミダゾールで溶出させることで分離を高めた(図3)。その後著者は、これら大腸菌由来の膜蛋白質が50mMイミダゾール存在下ではNi-NTA樹脂に吸着しにくいことを確認した。ただし、通常の6xHis蛋白質も吸着しにくい場合があるので10xHisタグは有効である。
培養について
前培養は本培養の1/20量のLB+0.5%グルコース培地で行う。本培養はTB培地で25℃、一晩で行う。IPTGは加えない。1 Lあたり、10–14 g湿重量の菌体が得られる。菌体は−80℃で保存する。
大腸菌にとって有害な蛋白質は前培養では発現させないほうがよい。そこで、T7 RNAポリメラーゼを発現させないように0.5%グルコース存在下で培養している。また、筆者はグルコースが発現制御だけでなくプラスミドコピー数の減少も引き起こしていると感じている。本培養ではIPTGを加えない。あまりにも大過剰な発現は正しくフォールドした蛋白質を与えず、活性を失っており、しかも可溶化できないことがある。
リポソームの調製について
クロロホルムを除いたあとガラス容器内壁に固着したリン脂質薄膜を、界面活性剤を含まないバッファーに懸濁し超音波処理で小胞化する方法も多用されている。粒子状の脂質をそのまま使用する場合は界面活性剤入りのバッファーで完全に溶融し、透析してもよい。
大腸菌リン脂質はフォスファチジルエタノールアミンが70%、フォスファチジルグリセロールが20%ほど含まれている。これに対して、ダイズやラットではフォスファチジルコリン、ついでフォスファチジルエタノールアミンが主成分である。蛋白質によって活性が変わることがあるので精製リン脂質の組み合わせを含め、適した脂質を選択するとよい。安価なダイズリン脂質(アゾレクチン)はアセトン洗浄して用いられている。
リン脂質を定量するには、酸分解ののち、無機リンの定量をする (5)。簡便法としては、1%OGを含むリン脂質溶液を超音波処理し、紫外吸収との関係式 \(\mathrm{[Abs\ 240\ nm]} = 0.4 \times \mathrm{[mg/mL\ Lipids]}\) を用いる方法がある(特開平5-66199、公開日 平成5年(1993)3月19日、キャノン株式会社)。
リポソームへの再構成について
本稿の条件では精製・濃縮して得られたChrSに含まれる持ち込みのDMと再構成時に加えた界面活性剤OGによって、ほぼ完全にリポソームが溶融分散している(膜蛋白質とリン脂質、界面活性剤の混合ミセル)。この状態から透析によって界面活性剤を除くことでプロテオリポソームが形成される。これに対して、界面活性剤を少量加え、脂質が溶融する寸前の状態(destabilized liposomes)にして膜蛋白質を加えてから界面活性剤を取り除く方法があり、ラクトース輸送蛋白質では高い輸送活性を保持したプロテオリポソームの調製を可能としている (6)。destabilized liposomesの生成は加えた界面活性剤がリポソームに浸潤することで生じる濁度上昇で確認することができる。加える膜蛋白質の量が多い場合は持ち込みの界面活性剤の寄与を無視できないので、膜蛋白質を加えてからの濁度変化を予め観察しておく。この方法でもChrSのヘム依存的自己リン酸化活性の回復が観察された。
膜蛋白質のリポソームへの組み込み効率はシュクロース密度勾配遠心による分画で評価できる (7)。膜蛋白質のリポソーム中での配向(どのくらい生体膜中と同じか、あるいは反転方向で埋め込まれているか)の評価は膜不透過性の基質を用いた酵素反応の測定や膜不透過性化学修飾剤による架橋、特異的抗体による標識などによって行うが、50 : 50という報告 (8) もあれば、一方向性のものもある (6)。再構成時に超音波処理を行うと両方向が混在するようである。ただし、著者は方向性を評価する実験法自体の信頼性(例えば用いた試薬がどの程度膜不透過性か?)に疑問を感じており、注意を要する実験であると思う。
界面活性剤の除去方法
リポソームやナノディスクへの再構成とは界面活性剤ミセルに覆われた膜蛋白質を再びリン脂質二重膜に埋め戻す操作である(図1、文献3)。この操作にはリン脂質存在下で界面活性剤を取り除く過程が含まれる。本稿のプロテオリポソーム編では透析法を用いたが、OGのような臨界ミセル濃度が極めて高いもの(0.7%)では希釈・超遠心法も有効である。また、ナノディスク編で述べているように、DDMやTX-100を用いた時には樹脂ビーズ(Bio-Beads SM-2)を加えて、界面活性剤を吸着させて取り除く方法もよく利用されている。
MSPについて
MSPは本来脂質と共存して安定化しているので、精製過程で凝集する場合がある。再構成実験では界面活性剤で可溶化して使用するので、凝集が生じたら予定している界面活性剤で溶かして使用または保存するとよい。
蛋白質定量では、Pierce社のBCA試薬ではAbs280nmによる定量値の1/2ほどの発色しか示さない(BioRadのRC DC Protein assayやCytoskeleton社Advanced Protein assay Reagentでは良好な結果を得た)。また、SDS-PAGE後の染色で和光純薬のQuick-CBBを用いたところ染色できなかった(バックグラウンドより先に脱色される)ので使用する試薬に注意を要する(従来のCoomassie brilliant blue R250染色やナカライテスク社CBB Stain Oneは使用できた)。
再構成したナノディスクについて
ChrSとリン脂質混合ミセルから界面活性剤を除いた場合には、超遠心操作によってほとんどすべてのChrSが沈殿する。これはリポソームに組込まれたものや凝集体の形成による。一方、MSPを共存させた場合には上清に回収されていることからナノディスクに再構成されていることがわかる(図9)。ChrSの濃度を変えることでMSPと等量的な再構成ナノディスクを得ることができる。
本研究で用いたナノディスクは直径が12nmであり、7回膜貫通ヘリックスをもつバクテリオロドプシン(bR)では3分子まで組込まれる(4,9)。ナノディスクの脂質層は面積8,900 Å2(片面のフォスファチジルコリン約150分子相当)あり、bR3分子は3,400 Å2(フォスファチジルコリン約60分子相当)を占めている。蛋白質へリックス一本につき断面積は140 Å2と概算でき、単純な計算ではあるが、脂質をまったく含まない場合へリックス60本ほどの面積である。実際の膜蛋白質には結合性の脂質が含まれている(例えばbR1分子に7–8個の脂質)。さらにナノディスク形成のためにMSPとの隙間を埋める脂質も必要であるため、組み込み可能な膜蛋白質のサイズはさらに小さいであろう。再構成時にChrSの量を変えて、得られたナノディスクのMSPとChrSの量をSDS-PAGEで調べたところ、6回膜貫通のChrSではホモダイマー1ないし2個が挿入されていることを示唆する結果を得た。
再構成ナノディスク中の膜蛋白質を定量するためには、簡易定量が可能なタグや色素が結合していない蛋白質の場合は、MSPとあわせ複数の既知量の精製標品と共にSDS-PAGEで展開し、染色バンドの濃淡を比較しなければならない。実際にスキャナーで染色ゲルの画像を取り込み、同一面積のバンドの濃淡を数値化したところ、数マイクログラム(100 pmol前後)で意外なほどの直線性が得られたので信頼性のある定量ができる。また、膜蛋白質が取込まれたナノディスクと空のナノディスクが混在するので、膜蛋白質固有のタグに依存したカラム精製などを行うことが望ましい。
参考文献
- Schmitt, M.P., J. Bacteriol, 181, 5330–40 (1999)
- Ito, Y. et al., FEBS Lett., 583, 2244–8 (2009)
- Rigaud, J.-L. & Lévy, D., Methods. Enzymol., 372, 65–86 (2003)
- Ritchie, T. K. et al., Methods. Enzymol., 464, 211–31 (2009)
- Boldog, T. et al., Methods. Enzymol., 423, 317–35 (2007)
- Knol, J. et al., J. Biol. Chem., 271, 15358–66 (1996)
- In’t Veld et al., Biochemistry, 31, 12493–9 (1992)
- Proteau et al., Biochem. J., 210, 199–05 (1983)
- Bayburt et al., Biochim. Biophys. Acta., 450, 215–22 (2006)
-
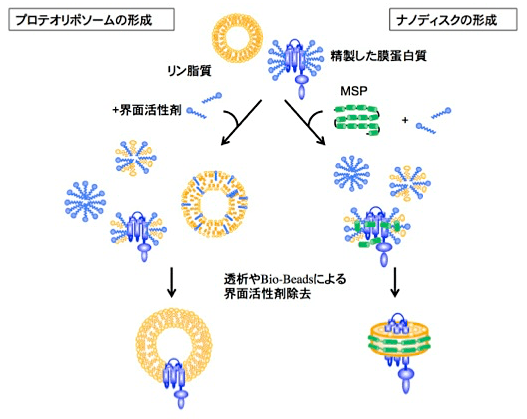
図1:プロテオリポソームとナノディスク形成による膜蛋白質の再構成 -

図2:デシルマルトシド、オクチルグルコシドで可溶化した大腸菌膜蛋白質
可溶化・超遠心後の上清をSDS-PAGEで展開した。ChrS蛋白質はオクチルグルコシドではほとんど可溶化できないことがわかる。 -
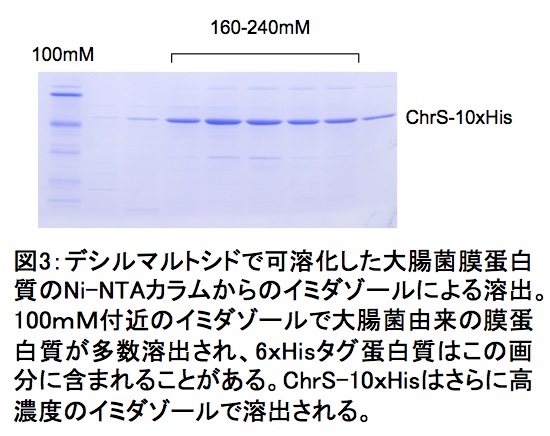
図3:デシルマルトシドで可溶化した大腸菌膜蛋白質のNiNTAカラムからのイミダゾールによる溶出
100 mM付近のイミダゾールで大腸菌由来の膜蛋白質が多数溶出され、6×Hisタグ蛋白質はこの画分に含まれることがある。ChrS–10×Hisはさらに高濃度のイミダゾールで溶出される。 -
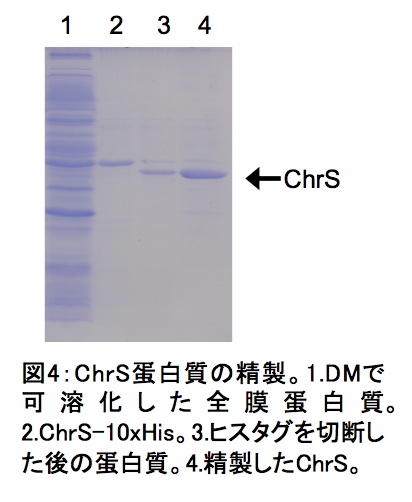
図4:ChrS蛋白質の精製 - DMで可溶化した全膜蛋白質。2. ChrS-10×His。3. ヒスタグを切断した後の蛋白質。4. 精製したChrS。
-
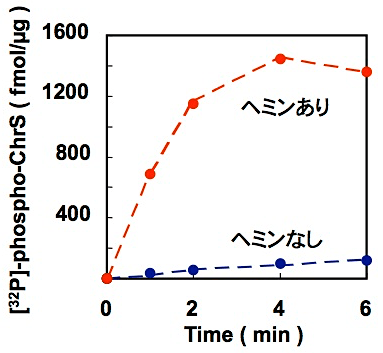
図5:リポソームに再構成したChrS蛋白質のヘム依存的自己リン酸化活性 -

図6:エッペンチューブ底に穴をあけるための怪我防止用自作ホルダー
段差付きくり抜き穴のあるゴムに再構成混合液、BioBeadsの入ったエッペンチューブを差し込み、逆さにして、チューブ底に針で穴をあける。 -

図7:バイオビーズ混合液から再構成液を遠心濾過分離するチューブ遠心後、17 mLチューブ底にたまった濾液を回収する。 -
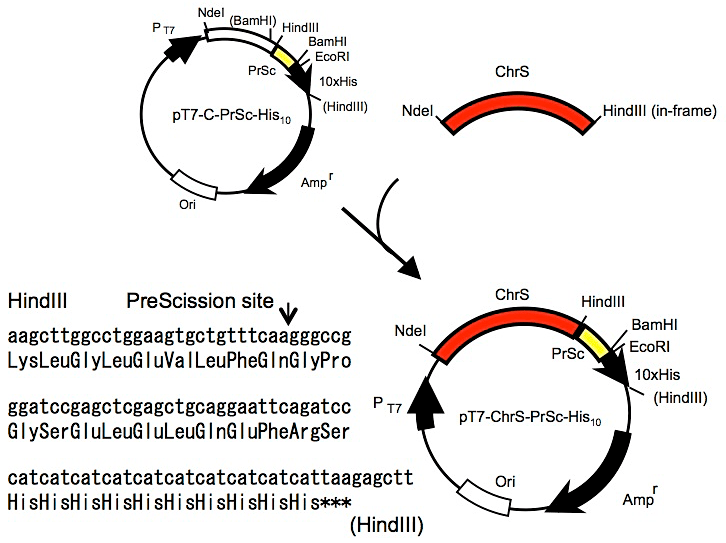
図8:ChrS発現プラスミドの作製とC末10×Hisの配列
膜蛋白質遺伝子のC末端にin-frameでHindIIIサイトを付加してある。 -
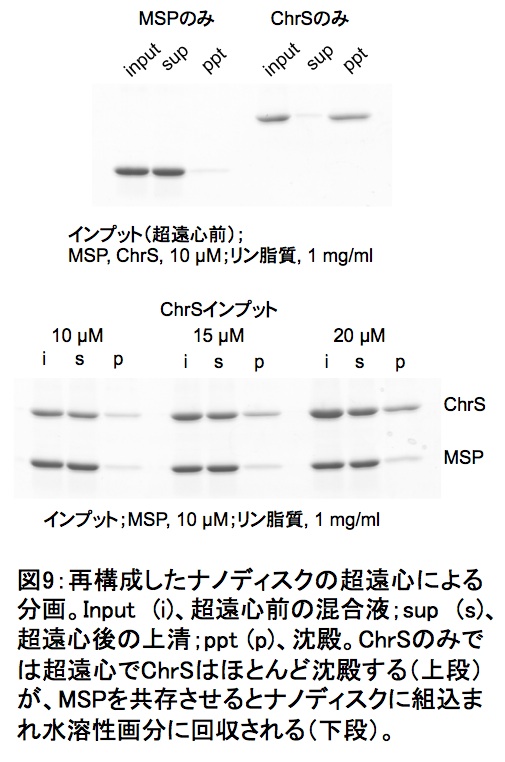
図9:再構成したナノディスクの超遠心による分画
Input(i)、超遠心前の混合液;sup(s)、超遠心後の上清;ppt(p)、沈殿。ChrSのみでは超遠心でChrSはほとんど沈殿する(上段)が、MSPを共存させるとナノディスクに組込まれ水溶性画分に回収される(下段)。
