

概要
アミロイドβ(Aβ)のNMR構造解析を行うためには、安定同位体標識を施したタンパク質が大量に必要となる。Aβのように凝集性の高いペプチドは、化学合成による調製がしばしば難しい。また、こうした短いペプチドをポリヒスチジン(His6)やグルタチオンSトランスフェラーゼ(GST)といった通常のアフィニティーカラム精製用タグ融合型で発現しようとしても、毒性による発現低下や、不溶性画分への移行により、その精製が困難である場合が多い。本プロトコールでは、ユビキチン融合型Aβの精製法および、スピンラベル化Aβの調製法について詳しく紹介する。本法は、Aβに限らず、他のペプチドの調製にも応用可能である。実際、我々はユビキチン融合タンパク質として、20~100残基ほどのペプチドおよび天然変性タンパク質の発現および精製に数多く成功している。
発現系の構築からAβの単離精製まで約2週間で完了する。
装置・器具・試薬
- 恒温振盪培養器、バッフルフラスコ
- 超音波破砕機
- 高速冷却遠心器
- 分光光度計
- オートクレーブ
- HPLC
- 凍結乾燥機
- 遠心エバポレーター
- pHメーター
- Ni-NTAカラム
- グルタチオンセファロース4B
- 逆相カラム
- カナマイシン
- アンピシリンナトリウム
- 尿素
- アセトニトリル(HPLCグレード)
- イミダゾール
- グルタチオン還元型
- AEBSF:4-(2-アミノエチル)ベンゼンスルフォニルフロライド
- EDTA:エチレンジアミン四酢酸
- DTT:ジチオスレイトール
- IPTG:イソプロピル-β-D(-)-チオガラクトピラノシド
- TFA:トリフルオロ酢酸
- Tris:トリスヒドロキシメチルアミノメタン
実験手順
1)ユビキチン融合型アミロイドβの大腸菌発現系の構築(数日~1週間)
2)ユビキチン融合型アミロイドβの培養・精製(2日)
3)脱ユビキチン酵素(YUH1)の培養・精製(2日)
4)ユビキチンタグの切断、アミロイドβの精製(1日~数日)
5)アミロイドβ溶液の調製(1日)
追記)スピンラベル化アミロイドβの精製(スピンラベル化反応~精製:2日)
実験の詳細
1)ユビキチン融合型アミロイドβの大腸菌発現系の構築
ユビキチン融合型ペプチド発現系の利点の一つとして、最終精製物として得られるペプチドのN末端に余分なアミノ酸残基が残らない点が挙げられる。通常のpETシステムやpGEXシステムを用いたHis6タグ融合タンパク質やGST融合タンパク質発現の場合、酵素が認識するアミノ酸配列の一部が切れ端として残る場合が多い。分子量の大きなタンパク質ではこのような切れ端の数残基の存在はしばしば無視されるが、短いペプチドの場合には一残基の付加によってその性質が大きく異なることは十分に予想される。また、本プロトコルでは、ユビキチンとAβを切断する脱ユビキチン酵素(YUH1)も大腸菌発現系により調製可能であり、低コストで効率よくAβを精製することができる。詳細は文献1-3を参照していただくとして、ここではポイントのみを説明する(図1)。
①His6タグ融合タンパク質発現用ベクターpET28a(Novagen)に、制限酵素BamHIおよびXhoIを用いてユビキチン(human)の遺伝子cDNAを組み込む。
②ユビキチンのC末端付近に変異を導入し、制限酵素AflIIの切断サイトを作製する。
③目的のペプチド(タンパク質)のN末端に、AflIIサイトとユビキチンのC末端配列を付加したプライマーを作製する。また、目的のペプチドのC末端の配列に終始コドンとXhoIサイトを付加したプライマーを作製する。これらプライマーを用い、PCRで増幅する。
④AflIIおよびXhoIサイトを用いて目的のペプチドをHis6タグユビキチンベクターに組み込むことにより、ユビキチンのC末端に目的のペプチドのN末端が直接連結したコンストラクトを作製する。
⑤得られたプラスミドのDNA配列を確認した後、大腸菌株 BL21-CodonPlus(DE3) (Stratagene)を形質転換する。
最終的に、脱ユビキチン酵素を用いてユビキチンのC末端を切断することにより(後述)、N末端に余分なアミノ酸残基が残らない目的ペプチドを調製することが可能である。
2)ユビキチン融合型アミロイドβの培養・精製
1. 準備
精製に使用する緩衝液の組成は下記の通りである。
- バッファーA:50 mM Tris-HCl(pH 8.0), 150 mM NaCl
- ソニケーション用:バッファーA + 1 mM AEBSF
- Ni-NTAカラム洗浄用1:バッファーA + 50 mM イミダゾール
- Ni-NTAカラム洗浄用2:バッファーA + 100 mM イミダゾール
- 溶出用:バッファーA + 500 mM イミダゾール
- 可溶化用:バッファーA + 8 M 尿素
- 透析用1:10 mM Tris-HCl(pH 8.0), 1 mM EDTA
- 透析用2:10 mM Tris-HCl(pH 8.0)
2. 前培養・本培養
前培養にはLB培地(5~10 ml)を用い、グリセロール中に保存した菌体懸濁液を少量加え、37 ℃で振盪培養を行う。本培養にLB培地を用いる際は、前培養した菌体溶液をそのまま全量添加する。一方、M9培地を用いる際は、前培養した菌体溶液を遠心分離(8,000 g, 10 min)し、得られた菌体を少量のM9培地で懸濁した後添加する。培養は37 ℃で行う。培養液のOD600が約1.0~1.2となった時点でIPTG溶液を終濃度 0.5 mMとなるように添加し、その後37 ℃で4~5時間培養を続ける。培養終了時のOD600はおおよそ1.5となる。培養終了後は、遠心し(8,000 g, 10 min, 4 ℃)、集菌する。
*ユビキチン融合型アミロイドβは可溶性画分と不溶性画分の両方に発現が認められる。封入体を可溶化することによりユビキチン融合型Aβを回収することが可能であり、かつ不溶性画分の方が大腸菌由来のプロテアーゼや夾雑物が少なく、精製効率が良い。したがって、誘導のタイミングは通常のタンパク質よりも遅めにしている。
3. 超音波破砕
ユビキチン融合型Aβ(1-40)を大量発現させた菌体をソニケーション用バッファー 35~40 ml(培養1 Lあたり)に懸濁し、50 mlビーカーへ移す。氷上で超音波処理を行った後、遠心分離(26,740 g, 30 min, 4 ℃)を行う。
上清画分:孔径0.45 μmシリンジフィルターにより脱粒子した後、Ni-NTAカラムへアプライする→4へ
沈殿画分:バッファーAに懸濁した後、遠心分離を再度行い、沈殿を回収する洗浄操作を2~3回繰り返す→5へ
4. 上清画分のNi-NTAカラムによる精製
Ni-NTAカラムとして、5 mLのchelating sepharose fast flow樹脂(GE Healthcare)にNi2+を配位させたレジンを用いる。
①Ni-NTAカラムはあらかじめ十分量(50 mL)のバッファーAで平衡化しておく。
②孔径0.45 μmシリンジフィルターにより脱粒子した上清画分を、平衡化したNi-NTAカラムへアプライする。
③バッファーA、Ni-NTAカラム洗浄用1バッファー、Ni-NTAカラム洗浄用2バッファーの順に十分量(50~100 mL)をカラムにアプライし、夾雑物をカラムから溶出させる。
④溶出用バッファーを40 ml添加し、ユビキチン融合型Aβ(1-40)を溶出する。精製したユビキチン融合型Aβ(1-40)の純度は、SDS-PAGE(15 %もしくは18 %)により確認する。
⑤溶出画分の透析を行う。2 L以上の透析用1バッファー、透析用2バッファーの順に、4 ℃にて各3時間以上行う。
⑥透析後、目的タンパク質の定量を行う。
*His6タグユビキチン融合型Aβ(1-40): M.W. 16364.5, ε(M-1,cm-1)= 2780
*ユビキチン融合型Aβ(1-40)の保存には、通常4 ℃を用いている。長期保存したい場合は、-80 ℃で凍結保存することも可能である。
5. 沈殿画分のNi-NTAカラムによる精製
可溶化:
沈殿画分に関しては、洗浄操作後、可溶化用バッファーを2~3 mlずつ添加し沈殿を可溶化する。沈殿が溶けるまで可溶化バッファーを添加する(最終体積:20 ml程度)。その後、遠心分離(26,740 g、30 min、4 ℃)を行い、得られた上清画分を孔径0.45μmシリンジフィルターにより脱粒子した後、Ni-NTAカラムにアプライする。
Niカラム精製:
Ni-NTAカラムとして5 mLのchelating sepharose fast flow樹脂(GE Healthcare)にNi2+を配位させたレジンを用いる。
①Ni-NTAカラムはあらかじめ十分量(50 mL)の可溶化用バッファーで平衡化しておく。
②孔径0.45 μmシリンジフィルターにより脱粒子した上清画分を、平衡化したNi-NTAカラムへアプライする。
③バッファーAを100 mLほどカラムにアプライし、夾雑物および尿素を溶出させる。
④Ni-NTAカラム洗浄用1バッファー、Ni-NTAカラム洗浄用2バッファーの順に十分量(50~100 mL)をカラムにアプライし、夾雑物をカラムから溶出させる。
⑤溶出用バッファーを40 ml添加し、ユビキチン融合型Aβ(1-40)を溶出する。精製したユビキチン融合型Aβ(1-40)の純度は、SDS-PAGE(15 %もしくは18 %)により確認する。
⑥溶出画分の透析を行う。2 L以上の透析用1バッファー、透析用2バッファーの順に、4 ℃にて各3時間以上行う。
⑦透析後、目的タンパク質の定量を行う。
3)脱ユビキチン酵素(YUH1)の培養・精製
ユビキチンからAβ(1-40)を切断する際に用いる脱ユビキチン酵素 YUH1を以下の手順で発現し精製する。
YUH1のN末端にGSTを融合させたタンパク質発現用ベクター(GST-YUH1)を用いる。
1. 準備
精製に使用する緩衝液の組成は下記の通りである。
- バッファーA:50 mM Tris-HCl(pH 8.0), 1 mM EDTA, 0.2 mM DTT
- 溶出用:バッファーA + 20 mM グルタチオン還元型
- 透析用1:50 mM Tris-HCl(pH 8.0), 1 mM EDTA, 0.2 mM DTT
- 透析用2:50 mM Tris-HCl(pH 8.0), 0.2 mM DTT
2. 前培養・本培養
前培養には50 μg/mL アンピシリンナトリウムを含むLB培地(5~10 ml)を用い、37℃で振盪培養を行う。前培養した菌体をLB培地に植菌し、25℃で本培養する。培養液のOD600が約0.7~0.8となった時点でIPTG溶液を終濃度が0.5 mMとなるように添加し、その後25℃で15時間培養を続ける。培養終了時のOD600はおおよそ1.2となる。培養終了後は、遠心し(8,000 g, 10 min, 4 ℃)、集菌する。
3. 超音波破砕およびグルタチオンカラムによる精製
①GST-YUH1を大量発現させた菌体をバッファーA 35~40 ml(培養1 Lあたり)に懸濁し、50 mlビーカーへ移す。氷上で超音波処理を行った後、遠心分離(26,740 g, 30 min, 4 ℃)を行い上清画分を得る。上清画分に終濃度が1 mMとなるようにDTTを添加する。
②グルタチオンカラムをあらかじめバッファーAで平衡化しておく。
③上清画分を孔径0.45 μmシリンジフィルターにより脱粒子した後、平衡化した15 mLのグルタチオンセファロース4Bカラム(GE Healthcare)にアプライする。
④280 nmにおける吸光度を指標としてタンパク質がほとんど流出されなくなるまでバッファーAを流す。
⑤溶出用バッファーを20~30 ml添加し、GST-YUH1を溶出させる。精製したGST-YUH1の純度は、SDS-PAGE(15 %)により確認する。
⑥溶出分画の透析を行う。2 L以上の透析用1バッファー、透析用2バッファーの順に、4 ℃にて各3時間以上行う。
⑦透析後の試料溶液に、終濃度2 mMとなるようにDTTを添加する。タンパク質の定量を行う。
*GST-YUH1: M.W. 52313.8, ε(M-1,cm-1)= 54050
⑧アミコンウルトラYM-10で限外濾過法により終濃度が0.8 mM程度になるようにGST-YUH1を濃縮する。濃縮後の試料溶液に、終濃度が2 mMとなるようにDTTを添加する。
*筆者は、すぐに使用する場合は4 ℃保存、長期保存する場合は-80 ℃で凍結保存している。
4)ユビキチンタグの切断・アミロイドβの精製
切断処理:
①Aβ(1-40)のN末端に融合しているユビキチンを切除するために、GST-YUH1のモル濃度をAβ(1-40)のモル濃度に対して1/10となるように添加する。終濃度が2 mMとなるようにDTTを添加した後、37 ℃にて1~2時間インキュベートする。
*一度に反応させる容量は、次の操作の逆相カラムへのアプライ量を考慮し決定する。通常、Aβ(1-40)溶液 6~10 mLとしている。
②切断反応の進行は、tricine-SDS-PAGEで確認する。
*tricine-SDS-PAGEに関しては、文献4を参考にしていただきたい。
逆相カラムによる精製:
HPLC精製に使用する溶液の組成は下記の通りである。
・A液:0.1 %(v/v) TFA
・B液:0.1 %(v/v) TFA + 80 %(v/v) アセトニトリル
*A液は、滅菌水1 Lを脱気した後、TFAを1 mL添加しよく撹拌する。
*B液は、滅菌水200 mLとアセトニトリル800 mLを別々に秤量し、混合した後、超音波洗浄器で20分脱気する。その後、TFAを1 mL添加しよく撹拌する。
HPLC精製は、流速1 mL/min、検出UV波長210 nmで行う。
①A液 98%、B液 2%で逆相カラムを平衡化する。
②切断処理を施した後の試料溶液に、A液を等量もしくは等量よりやや多めに添加してpHを3~4に調整する。
*Aβ(1-40)の等電点は5.2であるため、pHを下げる際にゆっくり行うと等電点付近で沈殿が生じてしまう。このため筆者は、A液の添加は一気に素早く行っている。等電点付近で白濁することがあるが、A液を添加してpHを下げることにより白濁は消失する。pHの確認は、pH試験紙で行う。
③孔径0.2 μmのシリンジフィルターに通して脱粒子した後、HPLCのループにインジェクトする。通常、5 mLのループを用いており、一度に4~4.5 mLの試料溶液をインジェクトする。
④A液でループ内の試料溶液を十分に逆相カラムへと流した後、B液を移動相としてグラジエントをかける。グラジエントはHPLC用B液を0分2 %(v/v)から40分で100 %(v/v)となるようにする。
⑤各ピークを分取する。
*はじめは0~40分でB液2 %~100 %(v/v)となるようなグラジエントで試し、Aβ(1-40)の分離が向上するように、カラムの種類に応じてグラジエントを工夫している。
⑥精製したAβ(1-40)の純度は、tricine-SDS-PAGEおよびMALDI-TOF-MS分析により確認する。
*アセトニトリルを除去するために、分取したサンプルの一部(100~200 μL)をマイクロチューブへ移し、エバポレーターで乾固させる。また、そのままの濃度ではクマシー染色で検出できない場合もあるので、乾固したサンプルに電気泳動のサンプルバッファーを10~20 μL添加することにより、10~20倍濃縮したサンプルを用いると良い。
⑦Aβ(1-40)を含む分画を回収し、凍結乾燥する。
*残存するTFA濃度を減らすため、滅菌水で希釈してから凍結乾燥している。
*完全に凍結乾燥した状態にすること。乾燥が不十分な場合、Aβ(1-40)溶液を調製する際に白濁・沈殿してしまうので注意する。
⑧凍結乾燥後のAβ(1-40)は使用時まで凍結保存(-20 ℃)する。
5)アミロイドβ溶液の調製
上記凍結乾燥後のAβ(1-40)粉末は直接バッファーには溶解されないので、最初にアルカリ溶液(0.1 %(v/v) NH3溶液)に高濃度で溶解した後、NMR測定用バッファーで希釈する必要がある。
Aβ(1-40)溶液の調製用(NMR測定用バッファー)として以下の組成の溶液を使用する。
- 10 mM リン酸カリウム [K2HPO4/KH2PO4](pH 7.2), 0.05 mM NaN3, 10 % D2O
①凍結乾燥後の試料に少量の0.1 %(v/v) NH3溶液を加えてすべて溶解した後、その一部を使用してBradford法によるAβ(1-40)溶液の定量を行う。
*NH3溶液の添加量が少ないと、Aβ(1-40)溶液が白濁する。その場合は、NH3溶液を素早く追加し、白濁が起こらない濃度まで希釈する。
*凍結乾燥が不十分な場合など、NH3溶液を添加しても白沈が残る場合がある。その場合は、遠心分離し上清のみ(通常、上から2/3ほど)を別のマイクロチューブに移し、使用する。
②必要量ごとにマイクロチューブに分注し、ストック溶液として使用時まで-30℃で凍結保存する。
③使用時は、Aβ(1-40)のストック溶液を氷上で解凍し、あらかじめ氷冷しておいたNMR測定用バッファーを加えてよく混和し、pH 7.2に調整する。最終的なAβ(1-40)溶液の濃度は測定に応じて0.05 mM~0.2 mMとする。
*実際には、10倍濃縮された溶液(100 mM リン酸カリウム, 0.5 mM NaN3)と、D2Oを別々に準備しておき、最終的に終濃度がNMR計測用バッファーの組成となるように体積を調整する。
*Aβ(1-40)のストック溶液は融解したら使い切ること。再凍結すると、再溶解した際に凝集し白濁する場合が多い。
追記)スピンラベル化アミロイドβの精製
ジスルフィド結合を介した部位特異的なスピンラベルを施すために、目的タンパク質にシステイン残基を導入することがしばしば行われる。Aβ(1-40)はシステイン残基を有していないので、遺伝子操作により特定のアミノ酸残基をシステイン残基に変異するか、N末端やC末端にシステイン残基を付加したコンストラクト[Cys-Aβ(1-40)]を作製する。筆者はN末端もしくはC末端にシステイン残基を付加したコンストラクトを作製し、NMR解析に用いた(5)。
システイン残基を付加したユビキチン融合型Aβ(1-40)の精製:
基本的には、野生型Aβ(1-40)の精製法と同じである。ただし、導入したシステインを介した分子間ジスルフィド結合の形成を抑制するために、Ni-NTAカラム精製用バッファーおよび透析用バッファーに0.2 mM DTTを添加する。また、透析後の試料溶液には終濃度で2 mMとなるようにDTTを添加する。
Aβの切断・精製:
野生型Aβ(1-40)の精製法と同様に行う。
スピンラベル化反応:
通常では、凍結乾燥させたCys-Aβ(1-40)を0.1 %(v/v) NH3溶液に溶解し、バッファーで希釈させたのちにスピンラベル化反応を行うが、DTTを除去するためにカラム精製を行う必要が生じる。この結果、Aβの凝集やカラムへの吸着などにより、収量が低下することがあった。そこで一連の操作をなんとか簡略化できないかと検討した結果、下記のプロトコールを確立することができた。これにより、精製過程を短縮することに成功し、収量を上げることに成功している。
①凍結乾燥後のCys-Aβ(1-40)をA液(0.1 %(v/v)TFA)に溶解後、アセトンに溶解したニトロキシドスピンラベル剤 MTSL(1-oxy-2,2,5,5-tetramethyl-D-pyrroline-3-methyl)-methanethiosulfonateをCys-Aβ(1-40)のモル濃度に対して5倍量添加し、4 ℃にて16時間反応させる。
*定量は凍結乾燥前にtricin-SDS-PAGEにより大まかに行っておく。
*MTSLはアセトンに10 mg/mLとなるように溶解する。アセトンに溶解後は-20 ℃で凍結保存する。
*スピンラベル化反応の進行は、MALDI-TOF-MS分析により確認する。反応が十分に進行していない場合は、MTSLを追加する。
②未反応のMTSLをサイズ排除クロマトグラフィー(PD-10 カラム、GE Healthcare)を用いて反応液から除去する。
③HPLCを用いて逆相クロマトグラフィーカラムによる精製を行う(図3)。
*HPLCの条件は、野生型Aβ(1-40)の精製法と同様。
*MTSL化されたCys-Aβ(1-40)と未反応のCys-Aβ(1-40)は溶出時間が異なるので、分離可能である。
④MTSL化されたCys-Aβ(1-40)を含む画分を回収し、凍結乾燥する。
⑤凍結乾燥後の試料は使用時まで凍結保存(-20 ℃)する。
工夫とコツ
沈殿分画からの可溶化および精製
ユビキチンは、尿素や塩酸グアニジンを用いて変性させても、それら変性剤を除くと容易に自発的に巻き戻る。したがって、通常のタンパク質を沈殿画分から精製する際に必要なリフォールディング操作を省略することができ、沈殿画分から高収率でユビキチン融合型Aβ(1-40)を得ることが可能である。さらに、沈殿画分由来の方が上清画分由来に比べて、大腸菌由来のプロテアーゼや夾雑物が少なく、HPLC精製を行う上で都合がよい。筆者は、Ni-NTAカラム精製までは上清画分と沈殿画分を同時に精製するが、沈殿画分により多くの目的タンパク質の発現を確認した場合には、沈殿画分を優先して精製している。上清画分に関しては、試しに1回分を切断処理・HPLC精製を行い、その結果に応じて精製するか否かを判断している。培養量を増やして沈殿画分からの精製量を増やした方が効率が良い場合もある。
ユビキチンタグの切断・アミロイドβの精製
ユビキチンタグが付加された状態のAβ(1-40)は安定であるが、ユビキチンタグを切断すると不安定性が増し、凝集しやすくなる。したがって、切断反応後は素早くHPLC精製を行っている。
ユビキチン融合型Aβ(1-40)をGST-YUH1で切断処理すると、Aβ(1-40)とHis6タグが付加したユビキチンに分かれる。したがって、この切断反応溶液をNi-NTAカラムに供することにより、His6タグ-ユビキチンのみを除去し、Aβ(1-40)を精製しようと試みたことがあるが、Aβ(1-40)がNi-NTAカラムに非特異的に吸着してしまい単離することができなかった。したがって、Aβ(1-40)を精製する際には、ユビキチンタグ切断後にNi-NTAカラムで精製することはお勧めしない。しかしながら、他のペプチド(Ni-NTAカラムに吸着しないペプチド)であれば、切断後にNi-NTAカラムに供することにより大半のユビキチンを除去することができ、後のHPLC精製が容易になる可能性があるので検討に値する。
逆相カラムの種類とグラジエント
同じような組成のカラムであっても、種類によってAβ(1-40)の溶出時間や溶出パターンが異なる。これまでに筆者が試したカラムとその用途をまとめた。カラムの種類や精製するペプチドに応じてグラジエントは各自で検討していただきたい。
A.TSKgel ODS-80TM, 4.6φ×250mm (TOSOH)
カラム長が長く分離が良いが、使用時の圧が他のカラムに比べて高くなる。また、分離が良い反面、夾雑物などもカラムによく吸着するため、カラムの劣化が早い。したがって、沈殿画分由来のAβ(1-40)やスピンラベル化したCys-Aβ(1-40)の精製に使用しており、上清画分由来のAβ(1-40)には使用していない。
*グラジエント例
| Time (mim) | 0 | 40 |
| B液(%) | 2 | 100 |
Aβ(1-40)は、24.3 min (B液:59.5%)あたりで溶出される(図2A)。
B.Mightysil RP-8 GP, 4.6φ×150mm(Kanto Chemical)
使用用途はODS-80TMと同様。分離能や吸着性もODS-80TMとほぼ同様。カラム長が短いためODS-80TMに比べると圧は低く、上清画分由来のAβ(1-40)にも使用している。
*グラジエント例
| Time (mim) | 0 | 1 | 16 | 17 |
| B液(%) | 2 | 35 | 62 | 100 |
Aβ(1-40)は、9.7 min (B液:50.7%)あたりで溶出される(図2B)。
C.Sunniest C8, 4.6φ×150mm (ChromaNic Technologies)
使用用途はMightysil RP-8 GPと同様。これまで試したカラムの中で、カラム圧・分離能・再現性が最も良く、最近はこのカラムを専ら使用している。沈殿画分由来のAβ(1-40)であれば、1回で単離精製が可能である。
*グラジエント例
| Time (mim) | 0 | 0.2 | 15 | 16 |
| B液(%) | 2 | 35 | 45 | 100 |
Aβ(1-40)は、11.7 min (B液:42.8%)あたりで溶出される(図2C)。
D.COSMOSIL 5C4-AR-300, 4.6φ×150mm(nacalai tesque)
カラムA~Cを用いた精製後に、Aβ(1-40)の分画に少量のユビキチンが混在している場合に、このカラムを用いて再度精製している。また、例えばAβ(1-35)や他のペプチドに関して、上記カラムA~Cを使用するとユビキチン由来のピークと完全に重なってしまい、分離できない場合には、このカラムを使用している。
*グラジエント例
| Time (mim) | 0 | 5 | 35 | 40 |
| B液(%) | 2 | 25 | 50 | 100 |
Aβ(1-40)は、26 min (B液:42.5%)あたりで溶出される(図2D)。
E.POROS R2/10, 4.6φ×100mm(Applied Biosystems)
Aβ(1-42)を精製するのに使用している。これまでの検討において、カラムA~DではAβ(1-42)がカラムに吸着してしまい溶出されなかった。カラム長が短く一度に多くのサンプルを打ち込むことができないため、精製に時間がかかるのが難点である。
*グラジエント例
| Time (mim) | 0 | 0.3 | 15 | 16 |
| B液(%) | 2 | 43 | 46 | 100 |
Aβ(1-42)は、6.6 min (B液:44.3%)あたりで溶出される。(図2E)
逆相カラムの取り扱いと洗浄
Ni-NTAカラム精製のみのユビキチン融合型Aβ(1-40)溶液にGST-YUH1を添加し、ユビキチンタグを切断した後、その反応溶液を直接HPLC(逆相カラム)へ打ち込むため、どうしても夾雑物の持ち込み量が多くなってしまう。少しでも夾雑物のカラムへの持ち込み量を減らすため、当然のことながらガードカラムを併用し、カラムへインジェクトする前に試料溶液を必ずフィルター(0.22μm)にかけること。
カラム圧が高い場合は、カラムの洗浄を行う。吸着物を溶出させるために、グラジエントをこまめに変える、100% アセトニトリル溶液を流す、カラムの溶出方向の逆向きから流す、などの処置を行う(各カラムの説明書に従って対応すること)。
カラムが劣化するとカラムの圧が非常に高くなり、洗浄しても下がらなくなる。そのような劣化したカラムでは分離が格段に悪くなるので、カラム交換をおすすめする。
文献
- Lee, EK., et al., Protein Expr. Purif., 40, 183-9 (2005)
- Utsumi, M., et al., Glycoconj. J., 26, 999-1006 (2009)
- Yagi-Utsumi, M., et al., Int. J. Alzheimers Des., 2011, 925073 (2011)
- 恩田真紀, 蛋白質科学会アーカイブ, 1, e011 (2008)
- Yagi-Utsumi, M., et al., FEBS Lett., 584, 831-6 (2010)
-
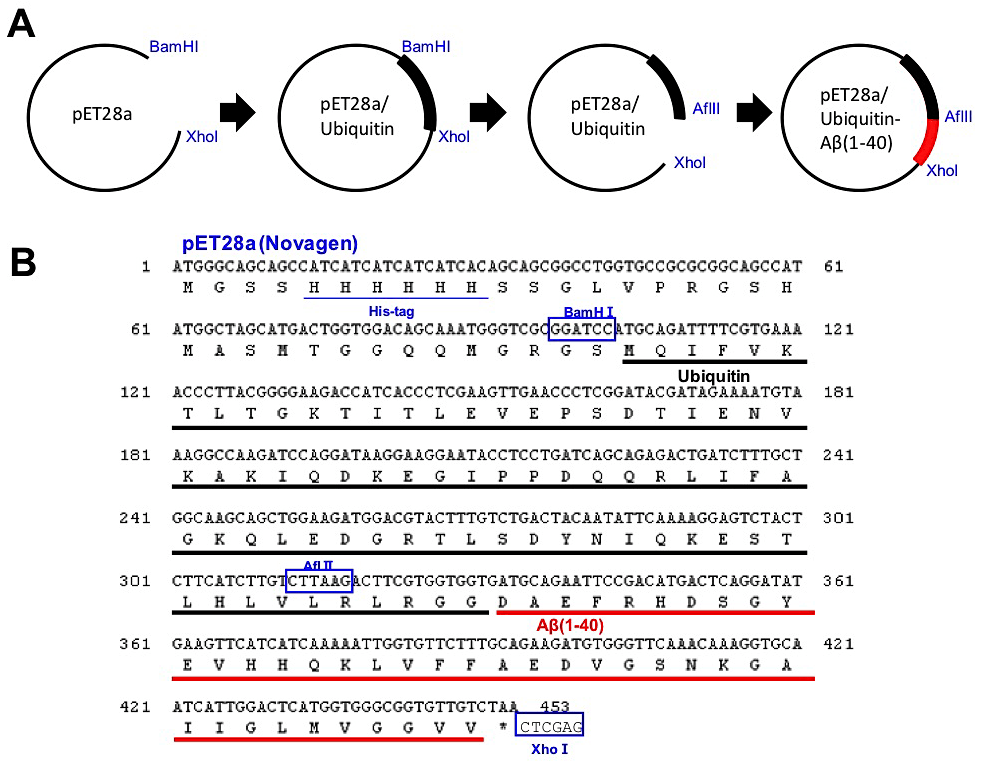
図1:ユビキチン融合型アミロイドβの大腸菌発現系の構築
(A)ユビキチン融合型Aβの発現プラスミドの作製の流れ、(B)pET28aベクターに組み込んだユビキチン融合型Aβのコドン配列およびアミノ酸配列。制限酵素サイトを青色四角で囲って示した。 -
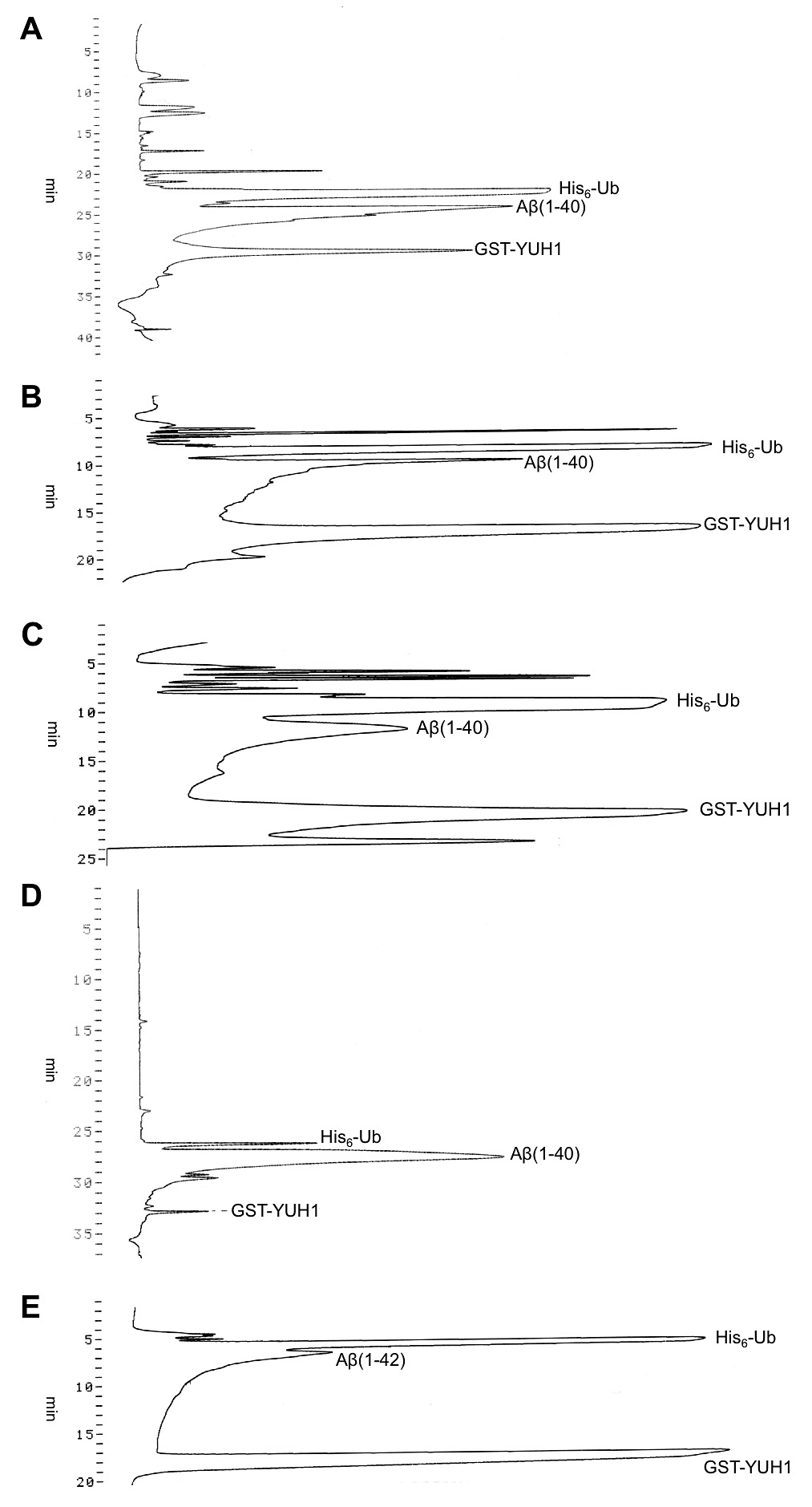
図2:逆相クロマトグラフィーによるAβ(1–40)およびAβ(1–42)の精製
各逆相カラム使用時のAβの溶出プロファイルを示した。(A)TSK gel ODS-80TM、(B)Mightvsil RP-8 GP、(C)Sunniest C8、(D)COSMOSIL 5C4-AR-300、(E)POROS R2/10 -
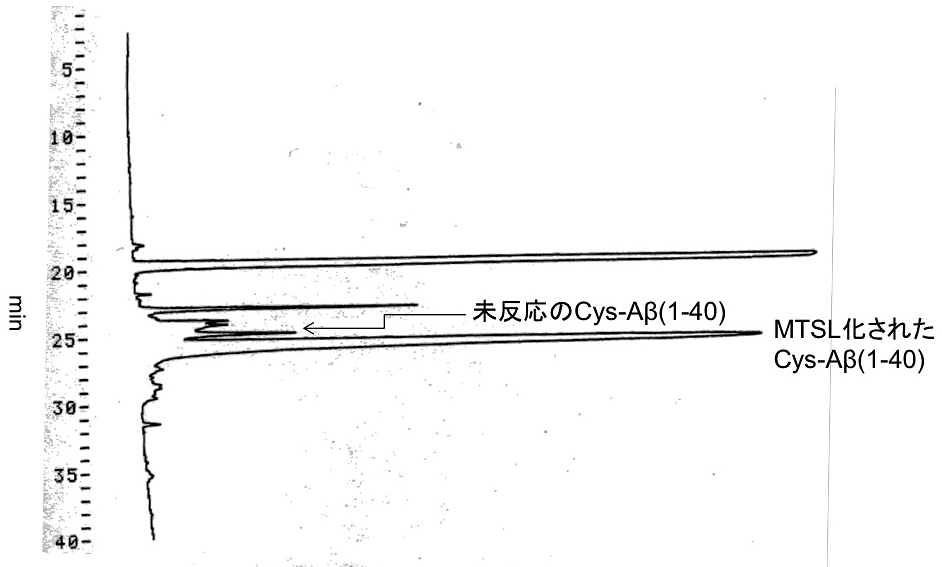
図3:逆相クロマトグラフィーによるスピンラベル化Cys-Aβ(1–40)の精製
逆相カラム(TSK gel ODS-80TM)使用時のAβの溶出プロファイルを示した。グラジエントはHPLC用B液を0分2%(v/v) から40分100%(v/v) とした。
