

概要
本稿では、出芽酵母(S. cerevisiae)を用いたGPCR改変体の迅速な作製・評価システムついて紹介する。目的タンパク質のDNA配列を、改変の導入に応じて断片化し、PCRにより増幅する。このPCR産物と発現ベクターを同時に用いて形質転換することで、S. cerevisiaeの細胞内で相同組換えにより環状の発現ベクターが構築される。形質転換株はそのままプレート培地上で選択可能で、引き続き少量培養を行うことで目的のタンパク質を発現させることができる。本システムではGFPをコードしたプラスミドを用いており、目的遺伝子を組込むことで発現させるタンパク質のC末端にGPFが融合される。それゆえ形質転換および少量培養後、続けてゲル内蛍光法、リガンド結合活性測定あるいは蛍光ゲル濾過法を行うことで、目的タンパク質の活性や物性を迅速に評価することができる。改変タンパク質の作製から評価までおよそ1週間と、昆虫細胞や動物細胞を用いるよりも大幅に短い期間で行うことができる。
イントロダクション
構造解析あるいは生化学的、物理学的解析を行う場合、目的のタンパク質を大量調製する必要がある。野生型の状態で安定に大量調製できるものは問題ないが、とくに高等生物のタンパク質は不安定で、異種宿主で発現した場合、発現量が低くその後の解析が困難となる。このような場合の対策として、タンパク質に改変を加えることで熱安定性を向上させたり、宿主での分解を低減させ安定に発現させたりするケースも多い。従来の方法では、大腸菌を用いて発現ベクター構築を行い、発現宿主に形質転換するため手間がかかった。GPCRの発現宿主としては昆虫細胞や、メタノール資化性酵母Pichia pastorisが用いられることが多い。よって従来は発現ベクターの作製から目的タンパク質の評価までおよそ3週間~1ヶ月程度要していた。
本稿で紹介するシステムでは、S. cerevisiaeの高い相同組換え活性を利用し、PCR断片と直鎖化した発現ベクターを同時に用いて形質転換することで、細胞内で発現ベクターが構築されるため、そのまま目的タンパク質の発現が可能である。また発現ベクターにはGFPをコードする領域が含まれ、目的タンパク質のC末端にGFPが融合する。よって発現した目的タンパク質を、GFPの蛍光を用いたゲル内蛍光法や蛍光ゲル濾過法によって解析することが可能であり、改変体の構築からタンパク質の評価までおよそ1週間で完結する。さらに酵母は大腸菌と同レベルの手法および施設で使用することができ、昆虫細胞や動物細胞よりも気軽にかつ安価に使用できるといったメリットがある。一方デメリットとしては、翻訳後修飾や細胞膜の脂質組成が動物細胞と異なり、哺乳類の膜タンパク質の発現能力に関して一般に昆虫細胞や動物細胞よりも劣ることなどが挙げられる(ただし改変体を作製することで克服できる場合もある)。
本稿では筆者らの論文 (1) を基に、筆者らの行っているプロトコルを記述した。本手法はDrewらの開発した手法 (2) を基盤としており、プロトコルとしては文献3も参考にして頂きたい。筆者らはGPCRをターゲットとして研究を進めているが、本システムは他の膜タンパク質や可溶性タンパク質にも利用可能である(可溶性タンパク質の例に関しては文献4を参照)。また、S. cerevisiaeの実験一般に関しては優れたプロトコル集があるのでそちらも参考にして頂きたい (5,6)。なお佐賀大学の永野先生のホームページに出芽酵母の相同組換えを利用したプラスミドの構築法について便利な記載がなされている。
装置・器具・試薬
装置
- サーマルサイクラー(各社)
- インキュベーター(プレート培地保温用)(各社)
- ブロックインキュベーター(1.5 mlチューブ用)(各社)
- 恒温震盪培養器(試験管用)(各社)
- 小型冷却遠心機(1.5 mlチューブ用)(各社)
- 冷却遠心機(&スイングローター)(各社)
※スイングローターの方が菌体の回収操作がしやすい。 - 蛍光プレートリーダー(各社)
- 細胞破砕装置(ガラスビーズによる破砕)(各社)
※VORTEX-GENIE2にターボミックスアタッチメントを付けるのが安価(図1A)。 - 位相差顕微鏡(各社)
- 小型超遠心機(& 1.5 mlチューブ用ローター)(各社)
- 蛍光イメージャー(GEヘルスケア社LAS-4010など)
- 中高圧クロマトグラフィー装置(BioRad社BioLogic Duo Flowなど)またはHPLC(各社)
- 蛍光検出器(島津製作所RF-20Aなど)→図1B、工夫とコツ(蛍光ゲル濾過法)
器具
- バイオリアクターチューブ(50 ml)(TPP社、BM機器カタログ番号87050)
※酵母の5~10 mlスケールの培養に用いる。バイオリアクターチューブは再利用不可である。1本100円程度と比較的高価であるため、現在筆者らはガラス製試験管とアルミキャップを用いている(下記)。 - ガラス製試験管(10 ml 培養用、サイズ24 mm × 200 mm)
- アルミキャップ(サイズ24.5 mm × 40 mm)
- 2.0 mlチューブ(各社)
- 超遠心用1.5 mlチューブ(Beckman社または日立工機社)
※各社の超遠心機に応じた純正のチューブを使うこと。 - 1.5 mlチューブ用ペレットペッスル(各社)
- ガラスビーズ(直径0.5 mm前後のもの)(各社)
S. cerevisiae FGY217株(遺伝子型;MATα ura3-52 lys2Δ201 pep4Δ)(2)
※ウラシル要求性であり、ウラシル欠損培地上では生育できない。また液胞の主要プロテアーゼである Pep4p を欠損しており、プロテアーゼ活性が抑えられている。
発現ベクターpDDGFP-2
※S. cerevisiaeの2μ複製起点をもつ高コピー型ベクターである。宿主のウラシル要求性を補填する選択マーカーであるURA3をもつ。ガラクトース誘導型のGAL1プロモーターによりタンパク質の発現が制御される。GAL1プロモーターの下流にGFPをコードし、目的タンパク質のC末端にGFPが付加される。ベクターの詳細は文献2を参照のこと。
試薬等
- 50%(w/v)グルコース溶液
※精製水に溶かし、フィルター滅菌。4℃保存。 - 25%(w/v)ガラクトース溶液
※Ura-培地(グルコース無し)に溶かし、フィルター滅菌。4℃保存。 - 1 M酢酸リチウム溶液
※オートクレーブ滅菌。 - 0.1 M酢酸リチウム溶液(1 Mの溶液を希釈して使用)
※オートクレーブ滅菌。 - 50%PEG3350
※PEG3350(Sigma社)を精製水に溶かし、フィルター滅菌 - Single-stranded carrier DNA溶液(2 mg/ml)
※High molecular weight DNA(Deoxyribonucleic acid sodium salt type III from salmon testes; Sigma社 D1626)100 mgをTE buffer 50 mlにスターラーで撹拌しながら溶かす(溶けるのに時間がかかる)。分注して−20℃保存。使用する時に95℃で5分間加熱し、その後氷中で急冷する。 - Yeast nitrogen base without amino acids & ammonium sulfate(BD社、233520)
- 硫酸アンモニウム(各社)
- Yeast Synthetic Drop-out Medium Supplements without uracil(Sigma、Y1501)
※自作可能(→工夫とコツ)。 - 制限酵素 SmaI(各社)
- プロテアーゼインヒビターカクテル(Roche社complete EDTA-free)
※1粒をMilliQ水1 mlに溶解し、50倍濃度の溶液を作製する。必要量を使用直前に加える。残った溶液は−20℃で1ヶ月程度保存可能。 - BenchmarkTM Fluorescent Protein Standard(Invitrogen社)
- 界面活性剤(各社)
- Superose6 10/300ゲル濾過クロマトグラフィーカラム(GEヘルスケア社)
Ura-培地(+2%グルコース、1 L)
| Yeast nitrogen base without amino acids & ammonium sulfate | 1.7 g |
| 硫酸アンモニウム | 5 g |
| Yeast synthetic drop-out supplement without Uracil | 1.92 g |
| 精製水 | 960 ml |
121℃、15分間オートクレーブ。
その後50%グルコース溶液を終濃度2%になるように(40 ml)加える。
※+0.1%グルコースの場合は、精製水を998 ml、50%グルコース溶液を2 ml加える。
実験手順
S. cerevisiaeはコロニーの成長に2~3日要するため、金曜日に形質転換を行い、土、日曜日をコロニー成長にあて、翌月曜日から少量培養を始めるように計画を立てるのが時間的なロスが少なくてよい。
- 準備
- 改変体の設計、プライマーの注文
- プラスミドの準備
- FGY217株のグリセロールストックをYPDプレートに起こす
- 形質転換
- 0日目(木曜日)FGY217株の植菌
- 1日目(金曜日)FGY217株の形質転換、目的遺伝子のPCR増幅(前もってPCRを行っておいてもよい)
- 2~3日目 プレートのインキュベーション
- 少量培養
- 4日目(月曜日)少量培養開始(形質転換株の植菌)
- 5日目(火曜日)少量培養(植継ぎ、インダクション)
- 6日目(水曜日)少量培養終了(菌回収、破砕、膜調製)
- 発現したタンパク質の評価
- 7日目(木曜日)ゲル内蛍光法、蛍光ゲル濾過法による目的タンパク質の評価
- 酵母からのプラスミドの抽出
実験の詳細
1)準備
改変体の設計
ここではあるGPCR改変体の作製例を挙げて、プライマー設計について説明する。このGPCRは比較的長いC末端と、非常に長いループ領域をもち、発現に悪影響を与えていそうなため、C末端は除去し、ループ領域には結晶化も念頭に安定なタンパク質であるT4リゾチームを挿入する。さらに安定化のために第3ヘリックスに部位特異的変異を導入する(図2A)。部位特異的変異導入の場合、オーバーラップする30塩基の中間に導入する(ここを中心として左右のTm値が近くなるような場所)。また異なるタンパク質を融合する場合は、融合するタンパク質のN末端とC末端に融合したい場所の配列を30塩基付加する(図2B)。この改変体の場合、1~8のプライマーを設計し注文する。オーバーラップする領域を同じ色で示している。
プラスミドの準備
発現ベクターpDDGFP-2を制限酵素SmaIでカットしておく。まず各社SmaIに応じた組成でpDDGFP-2を消化する。これを60℃、20分間加熱し酵素を失活させる。そして滅菌水でプラスミドの終濃度が30~40 μg/μlとなるように希釈する。調製したSmaI-cutプラスミド溶液は、凍結融解の繰返しを避けるため小分けして、−20℃で保存する(10回程度の凍結融解は問題無い)。
※S. cerevisiaeの形質転換効率が高いため、少量のプラスミドで十分なコロニー数を獲得できる。筆者らは1回(5 ml)の Miniprep でおよそ30 μgのpDDGFP-2プラスミドを得ているが、これは800~1,000回分の形質転換に使える量である。
FGY217株をYPDプレートに起こす
使用するS. cerevisiae FGY217株を、グリセロールストックからYPDプレートに起こしておく。グリセロールストックを-80℃から出し、滅菌したイエローチップの先で凍っているところを少し削り、YPD培地に塗布する。残りはすぐに-80℃に戻す。YPDプレートを2~3日間、30℃でインキュベートする。コロニーが成長したら4℃で保存する。起こした菌は2ヶ月間程度は問題なく使える。
2)形質転換
0日目 FGY217株の植菌
YPDプレートから爪楊枝でFGY217株を掻きとり、YPD培地5 mlに植菌する。30℃で一晩振とう培養する。
1日目 目的遺伝子のPCR、FGY217株の形質転換
目的遺伝子のPCR
改変体作製のためのPCRを行う。PCRに使用するポリメラーゼはフィデリティーの高いものを用いる(Roche社のExpand high fidelity、TOYOBO社のKOD plusなど)。反応液は25 μlあれば十分である。各社PCRシステムのマニュアルに従って反応液を調製しPCRを行う。PCR終了後、反応液を1 μlとり、アガロースゲル電気泳動により増幅を確認する。
FGY217株の形質転換(→工夫とコツ)
- (朝)滅菌水で20倍希釈しODをチェックする。OD600 = 10ぐらいになっている。
- 滅菌した200 ml三角フラスコにYPD培地を50 ml入れ、OD600 = 0.12になるように培養液を植え継ぎ、30℃で振とう培養する(50 mlで約20サンプルの形質転換が可能である)
- OD600 = 0.6~0.8になるまで培養する(5時間程度)。
(振とう速度は、往復型振とう機であれば130~150 rpm/min、回転型であれば200~250 rpm/min。いずれにしても酵母がそこに溜まらない程度の速度)
※この間にPCRやSingle-stranded carrier DNA溶液の加熱、急速冷却を行う。 - 培養液を50 ml遠沈管に移して、スイングローターで3,000 g、5分間遠心し、上清をデカントで捨てる。
- 菌体(沈殿)を滅菌水25 mlに懸濁する。
- スイングローターで3,000 g、5分間遠心し、上清をデカントで捨てる。
- スイングローターで3,000 g、5分間再度遠心し、上清をピペットマンできれいにとる。
- 0.1 M LiAc 1 mlに懸濁。1.5 mlチューブに移す。
- 12,000 gで5秒間遠心。上清を捨てる。
- 0.1 M LiAc 1 mlに懸濁する。
※培養液50 mlの場合の量である。培養液の量に応じて適宜0.1 M LiAcの量を変える。 - 滅菌した1.5 mlチューブに以下の物を順に加える。
- SmaI-cut pDDGFP-2 1 μl
- PCR産物 各3 μl
- 滅菌水 ○○ μl(この段階で50 μlになるように)
- Single-Stranded Carrier DNA溶液 25 μl
- 50%PEG3350 240 μl
- 1.0M LiAc 36 μl
全て加えたらすぐにボルテクスでよく混ぜる。
- 10で懸濁した菌液を50 μlずつ11のチューブに入れる。菌液を入れたらすぐにボルテクスでよく混ぜる。
- 30℃、30分間インキュベート
- 42℃、20~25分間ヒートショック
- 8,000 gで15秒間遠心、上清を除く。
- 0.2 mlの滅菌水で静かに懸濁する
- Ura-プレート培地に適当量まき(50μl程度)、30℃で2~3日間インキュベーション。コロニーが大きくなってきたら4℃で保存。
2~3日目 プレートのインキュベション
3)少量培養
4日目 少量培養開始(形質転換株の植菌)
コロニーをUra-培地(+2%グルコース)3 mlに植菌する。30℃、O/N振とう培養する。(→工夫とコツ)
※増殖が遅めなので、スケジュール上、昼頃までにはやった方がよい。
5日目 少量培養(植継ぎ、インダクション)
- (朝)20倍希釈してOD600チェック(通常7~8ぐらいになっている)
- OD600 = 0.12になるように、Ura-培地(+0.1%グルコース)10 mlに植え継ぐ。
- OD600 = 0.6ぐらいになるまで、30℃、6~7時間振とう培養する。
- OD600をチェック(培養液を0.8 mlとり、そのまま測定)。Ura-培地で調製した25%ガラストースを、ガラストースの終濃度が2%となるように入れる(約850 μl)。グリセロールやDMSOなどを培地に添加する時はここで添加する。
- 30℃で20~22時間振とう培養する。20℃で培養する時はこの時点から20℃に下げ、38~40時間培養する。
※残りの菌液を使って形質転換株保存用のコロニープレートを作製する。残った菌液を2 μl程度Ura-プレートに落とし、フタを上にして乾燥するまで待つ。乾燥したらひっくり返し、30℃でインキュベートする。2日間程度で菌がこんもりとなるぐらいに成長する。4℃で1~2ヶ月間保存可能。
6日目 少量培養終了(菌回収、破砕、膜調製)
- 20倍希釈でOD600をチェックする。(省略してもよい。培養条件や発現量によりODは異なるが、OD600=4~7ぐらいになっている。)
- 培養液を15 ml遠沈管に移し、スイングローターで3,000 g、5分間遠心。上清をデカントで捨てる。
- 3,000 g、1分間遠心。上清をピペットマンを使ってきれいに取り除く。
- 菌体をSuspension buffer(50 mM Tris-HCl pH7.5、5 mM EDTA、10%グリセロール、0.12 Mソルビトール、1x Complete EDTA-free)700 μlに懸濁し、2 mlチューブに移す。
- 懸濁液をよくボルテクスしたのち、20 μlをとって180 μlのSuspension bufferで希釈し、蛍光プレートリーダーでGFPの蛍光強度を測る。
- 懸濁液に500 μlのグラスビーズを入れ、チューブのキャップにパラフィルムを巻く。
- ボルテックスでMax Speed、10分間破砕(コールドルームで行う)。顕微鏡で破砕具合を確認する(図3)
※8割以上破砕できていればよい。 - 12,000 gで1分間遠心し、未破砕の菌と破砕後のデブリを落とす。
- 上清500 μlを超遠心用のチューブに移す。
※グラスビーズを取らないように注意。 - 100,000 g、30分間超遠心し膜画分を集める。
- 上清をピペットマンできれいに除く。
- 膜画分をMembrane buffer(50 mM Tris-HCl pH7.5、120 mM NaCl、20%グリセロール、1x complete EDTA-free)50 μlに懸濁する。ペレットペッスルを用いてよく懸濁する。→膜懸濁液
※膜懸濁液は一晩程度ならば氷中に置くことができるが、長期の場合は−80℃で保存する。
4)発現したタンパク質の評価
7日目 ゲル内蛍光法、蛍光ゲル濾過法による目的タンパク質の評価
ゲル内蛍光法
ゲル内蛍光法は、SDS-PAGEのゲル中でGFPが蛍光を発することを利用し、目的タンパク質のバンドを簡便に見ることができる方法である (8)。ウェスタンブロッティングのような煩雑な操作を必要とせず、便利である。図4に筆者らの例を載せた。
- SDS-PAGE 用のゲルを作製する。
※ トリス–グリシン系で行う。それ以外ではGFPの蛍光が見られないことがある。 - 膜懸濁液10 μlに、2x SDS-PAGE sample buffer(50 mM Tris-HCl pH7.5、5 mM EDTA、5%グリセロール、4%SDS、0.02%BPB、50 mM DTT、1x complete EDTA-free)を10 μl加える。室温に20~30分間置き、ゲルに10 μlアプライする。サンプルをボイルしてはいけない。マーカーは Benchmark™ Fluorescent Protein Standard(Invitrogen社)を1~2 μlアプライする。
※ 2x SDS-PAGE sample bufferと混ぜて長時間おくと、GFPは徐々に変性し蛍光が失われる。遅くとも1時間以内には泳動するのがよい。 - 泳動が終了したら、精製水を入れた容器にゲルを移す。
- 蛍光イメージャー(LAS-4010など)を用いて、青色光源でY515フィルターを通して撮影する。
蛍光ゲル濾過(FSEC)法
蛍光ゲル濾過法は、目的タンパク質に融合したGFPの蛍光を利用することで、目的タンパク質由来のゲル濾過クロマトグラムをクルードな状態でも検出することができ、簡便にその性質を知ることができる便利な方法である (9)。詳しくは蛋白質科学会アーカイブ, 3, e057 (2010)「GFPタグを利用した原核生物由来膜タンパク質の発現系評価」を参照して頂きたい。ここでは筆者らが行っている手順を記す。→工夫とコツ
- ゲル濾過カラムを、Gel filtration buffer(20 mM Tris-HCl pH7.5、150 mM NaCl、0.03%DDM/0.006%CHS)で平衡化しておく。
- 膜懸濁液30 μlに、可溶化Buffer(50 mM Tris-HCl pH7.5、200 mM NaCl、1%DDM/0.2%CHS、1x complete EDTA-free)170 μlを加える。
※可溶化の組成は適宜変えること。 - 4℃で1時間撹拌し可溶化する。
- 100,000 g、30分間超遠心する。
- 上清を別のチューブに移す。
- 100 μlをインジェクトし分析する。
5)酵母からのプラスミドの抽出
- 形質転換酵母をUra-培地(+2%グルコース)5 mlに植菌し、30℃でO/N震盪培養。
- 1.5 mlチューブに集菌する。
- Sorbitol buffer(1 Mソルビトール、0.1 M EDTA pH7.5)500 μlに懸濁する。
- 12.5 mg/ml Zymolyase-20T(ナカライテスク)を20 μl加え、37℃、30分インキュベート。
※Zymolyase 溶液は用事調製。 - 12,000 g、1分遠心、上清を除く。
- QIAprep Miniprep Kit(Qiagen社)のBuffer P1 250 μlで懸濁する。
- Acid-washed glass beads (Sigma社G8772)を150 μl加える。
- 1分間 Maxスピードでボルテックス
- Buffer P2を250 μl加え、以下キットのプロトコルに従う。
- 大腸菌に形質転換し、プラスミドを増やす。→シーケンス確認
※プラスミド抽出キットは他社のものでも良い。
工夫とコツ
どのような改変体を設計するか
野生型では発現が低くても、改変を施すことで劇的に発現が向上するタンパク質も多い。どのような改変を施すかはタンパク質によって様々である。GPCRの場合、フレキシビリティーの高いN末端、C末端や細胞内第3ループを削除したり、そこに別の安定なタンパク質を融合したりことで発現が向上するケースが多い。また熱安定性の高い試料調製は、構造解析、生化学的解析を行う上で重要である。GPCRにおいては、部位特異的変異導入 (10,11) や安定なタンパク質の融合 (12) により熱安定性が向上するケースが報告されている。膜タンパク質の熱安定性評価法としては蛍光色素を用いる方法 (13)、RI標識リガンドを用いた結合試験による方法 (11)、蛍光ゲル濾過による方法 (14) が報告されている。
Yeast Synthetic Drop-out Medium Supplements について
特定の成分を含まないDrop-out SupplementがSigma社などより発売されているが、高価である。よってたくさん実験する場合は自作するのがよい。含まれる構成成分は各Drop-out Supplementのマニュアル(例えばSigma社Y1501など)に詳細に記述されており、ダウンロード可能である。自作する場合、各構成成分を乳鉢に入れ、丹念にすりつぶしながら均一になるように混ぜる。これを乾燥した遮光性の試薬瓶に入れ、デシケーターの中で保管する。時間が経つと白→薄褐色に変色してくるが問題なく使えている。
酵母の植菌
大きめのコロニーを、爪楊枝を使って掻き取る。増殖速度が大腸菌よりも遅いため、爪楊枝の先に酵母が乗っているのが目視できるぐらいの量を掻き取り、試験管培地に良く懸濁する。爪楊枝に含まれる成分が酵母の生育を阻害する場合があるため、試験管に爪楊枝を投入しない方が良い。未消化のプラスミドを取り込んだりPCRのエラーにより目的のタンパク質を発現しないコロニーに当たることもあるので、通常1つの改変体につき2個以上のコロニーの発現をチェックする。
形質転換
DNA断片の数が多いほど形質転換効率は落ちる (15)。断片5つぐらいならば問題なく行うことができる(それ以上も可能だとは思うが筆者は試したことがない)。
初めてSmaI処理したプラスミドを作ったときは、PCRの断片の代わりに水を入れたもの(ネガコン)を一緒にトランスフォーメーションする。通常、ネガコンのコロニー数は5%より少ないが、ネガコンのコロニーが多いとその後の実験に支障が出る。このような場合は酵素消化に問題があるといえる。
発現したタンパク質の評価
本稿では発現したタンパク質の評価法として、ゲル内蛍光法と蛍光ゲル濾過法を挙げた。ゲル内蛍光法は発現タンパク質の完全性(integrity;全長発現しているか、分解されていないかなど)をチェックする方法として有用である。また蛍光ゲル濾過法は実際に界面活性剤で可溶化した際の挙動が分かるので、サンプルが精製まで持って行けるかどうかを判断する重要な指標となる。一般には目的の分子量付近にシャープで対象なピークが得られれば「良好」であると判断できる。図5に筆者らが行った蛍光ゲル濾過法の例を示す。この他にも受容体や輸送体であればその活性を見るということも考えられる。またDrewらは共焦点レーザー顕微鏡を用いて発現した膜タンパク質の局在を見ることで、きちんと発現しているかどうかある程度検討をつけることができるとしている (2)。どの程度の熱安定性が必要かということに関しては、熱安定性の測定方法、結晶化方法により異なるため一概に言うことはできない。筆者らが構造解析を行ったGPCRに関しては、蛍光色素(CPM)を用いた方法で、Tm値がおおよそ50℃程度であった。
蛍光ゲル濾過法
HPLCシステムはもちろんのこと、既存の中圧クロマトグラフィーシステムに蛍光検出器をつなげて使うこともできる。筆者らはBioLogic Duo Flow(BioRad社)に蛍光検出器(島津製作所)をつなげて使っている(図1B)。この場合、蛍光検出器のシグナルをBioLogicのシステムに取り込むためのシグナルインポートモジュール(BioRad社、10万円ぐらい)が別途必要である。
また設備が無い場合は、96ウェルプレートにフラクションを取り、蛍光プレートリーダーで読んでグラフ化しても同様のクロマトグラムが描ける(文献1、2、16ではこの方法をとっている)。その場合100 ml程度の培養が必要である。
界面活性剤の選択
GPCRの場合、多くはドデシルマルトシド(DDM)にコレステロールヘミコハク酸(CHS)を混ぜたものを用いることが多い。目的の膜タンパク質に適した界面活性剤を選択する必要がある。
性質の良い改変体が得られたら
野生型より発現量が高く、蛍光ゲル濾過法での単分散性の良い改変体が得られたら、まずはそのままS. cerevisiaeの形質転換株を大量培養し、精製を試みるとよい。本稿の主題は、S. cerevisiaeを用いた改変体の作製および評価であるので、これより後のことは記述しない。S. cerevisiae-GFPの系を用いた大量培養および精製に関しては、Drewらの文献に詳しい (2,3,16)。
一般に培養1 Lあたり最終精製物が0.1 mg以上得られれば「良好」であるといえる。野生型よりも発現量、単分散性が良くなったが、S. cerevisiaeではまだ目的の発現量に達しないような場合は、P. pastorisに、または施設が十分に調っていれば昆虫細胞に発現宿主を移すことで、さらなる大量発現が見込める場合もある。筆者らはヒト由来ヒスタミンH1受容体について、S. cerevisiaeを用いて改変体を作製し大量調製を試みたが、宿主をP. pastorisに移すことでさらなる大量発現が可能となった (1)。
文献
- Shiroishi M. et al., Microb. Cell. Fact., 11, 78 (2012)
- Newstead S. et al., Proc. Natl. Acad. Sci. USA, 104, 13936-41 (2007)
- Drew D. et al., Nat. Protoc., 3, 784-98 (2008)
- Ito et al., Biotechnol. Lett., 33, 103-107 (2011)
- 大矢禎一監訳 酵母遺伝子実験マニュアル,丸善 (2002)
- 水野貴之 バイオ実験イラストレイテッド7 使おう酵母できるTwo Hybrid, 秀潤社 (2003)
- Kota J. et al., J. Cell Biol., 176, 617-28 (2007)
- Drew D. et al., Nat. Meth.,3, 303-313 (2006)
- Kawate et al., Structure, 14, 673-681 (2006)
- Roth C.B. et al., J. Mol. Biol., 376, 1305-1319 (2008)
- Serrano-Vega M.J. et al., Proc. Natl. Acad. Sci. USA, 105, 877-882 (2008)
- Chun E. et al., Structure, 20, 967-974 (2012)
- Alexandrov A.I. et al., Structure, 16, 351-359 (2008)
- Hattori M. et al., Structure, 20, 1293-1299 (2012)
- Ito K. et al., Biochem. Biophys. Res. Commun., 371, 841-845 (2008)
- Sonoda Y. et al., Structure, 19, 17-25 (2011)
-

図1:使用する装置
A:酵母の細胞破砕に用いている装置。B:BioLogicに蛍光検出器をつないで蛍光ゲル濾過を行う図。筆者らはチャンバーの中にスペースがなかったので、蛍光検出器を外に置きチャンバーに穴を開けてつないでいる -
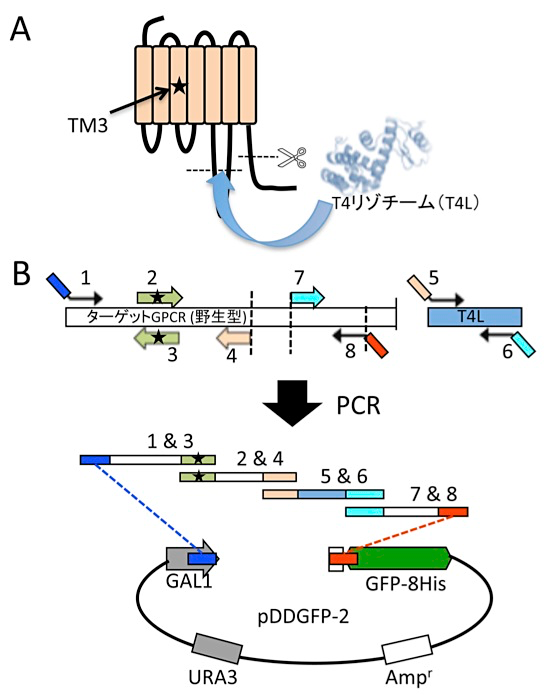
図2:A:改変体の例、B:プライマーの設計 -
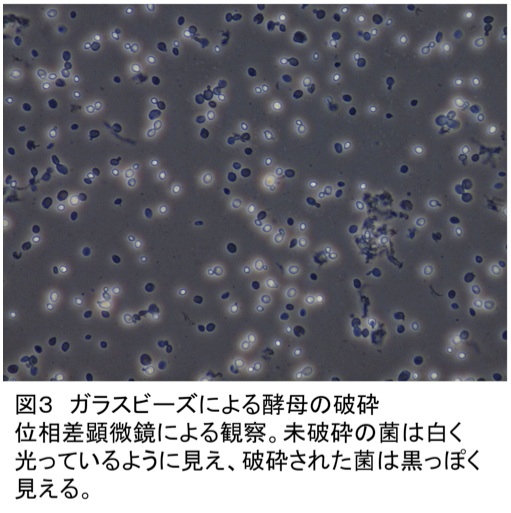
図3:ガラスビーズによる酵母の破砕
位相差顕微鏡による観察。未破砕の菌は白く光っているように見え、破砕された菌は黒っぽく見える。 -
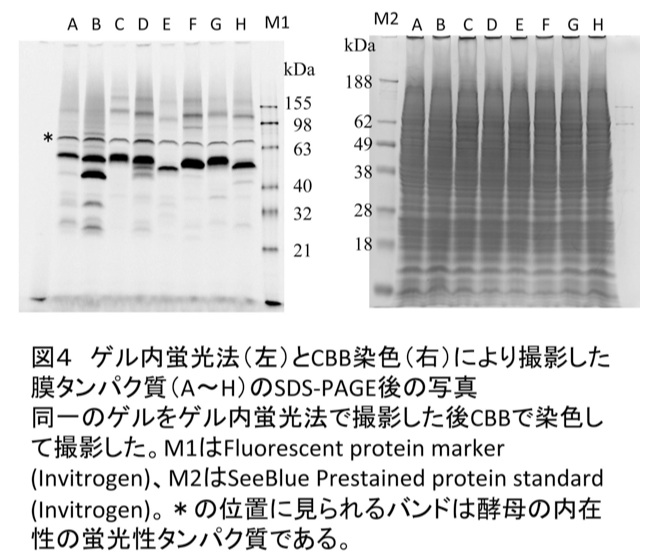
図4:ゲル内蛍光法(左)とCBB染色(右)により撮影した膜タンパク質(A~H)のSDS-PAGE後の写真
同一のゲルをゲル内蛍光法で撮影した後CBBで染色して撮影した。M1はFluorescent protein marker(Invitrogen)、M2はSeeBlue Prestained protein standard(Invitrogen)。*の位置に見られるバンドは酵母の内在性の蛍光性タンパク質である。 -
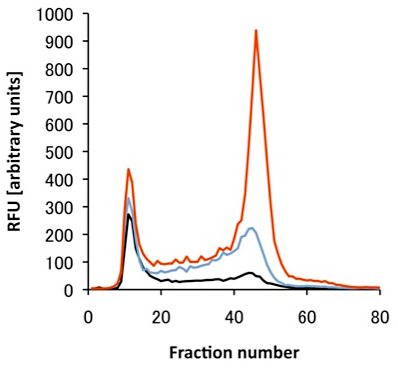
図5:蛍光ゲル濾過法による解析例
あるGPCRについて行った、野生型(黒)、改変体1(水色)、改変体2(赤)の蛍光ゲル濾過クロマトグラム。カラムはSuperose6 10/300を用い、96ウェルプレートにフラクションを分取し、蛍光プレートリーダーでGFPの蛍光を測定した。改変体2については大量精製に成功している。
