

概要
メタノール資化酵母Pichia pastoris(以下ピキア)を用いた組み換えタンパク質発現の特徴と手順について述べる。おおざっぱに言うとピキアを用いた発現系は大腸菌のそれと、昆虫細胞や動物細胞のそれの間に位置するものである。つまり、発現系構築の手間や培養時間は動物細胞ほどではないが大腸菌よりはかかる。一方、タンパク質の翻訳後修飾は動物細胞のようにはされないが、大腸菌のように翻訳されっぱなしではなくちゃんと折り畳んだ状態で発現する、という感じである。こういった特徴は、ピキアは単細胞生物ではあるが真核生物である、というところから来る。もし大腸菌の発現系で目的のタンパク質がちゃんと折り畳んだ状態で発現しなかった場合、ピキア発現系を試してみるのも一案だと思う。まず簡単にピキア発現系の利点、欠点を列挙してみる。
利点
- 発現タンパク質のN末端側にαファクターのprepro配列という酵母特有のシグナル配列を導入することで、発現タンパク質を細胞外の培地中に分泌させることができる。このことにより、細胞破砕等をする必要がなく、精製が比較的楽になる。
- 細胞外に分泌される過程で様々なシャペロンタンパク質の影響を受けて、ちゃんと折り畳まった状態で目的のタンパク質が分泌される。そのため、発現後のリフォールディング操作をする必要が少ない。特にジスルフィド結合を複数持ったタンパク質の場合、その効果は絶大である。
- 目的タンパク質によっては発現量が非常に高くなる。(筆者の最高記録はあるβラクトグロブリン変異体で1 g/L培地、平均で約100 mg/L)
- NMR測定サンプル用の同位体ラベルをする系が確立している。
欠点
- 目的タンパク質のN末端に余分なアミノ酸が付加してしまう場合がある。しかも場合によってはその残りの配列が不均一になる。シグナル配列全部が完全に取り除かれないからである。
- 高等生物のような翻訳後修飾(リン酸化など)はされない。糖鎖付加はされるが他の生物とは異なった糖鎖が付加されてしまう。
- ジャーファーメンターがある方が良い。
筆者の所属していた研究室ではウシβラクトグロブリン (1) やヒトβ2ミクログロブリン (2) といったタンパク質を発現してきた。本稿ではそれらのタンパク質の発現を行った時の手順を紹介する。なお、近年のピキア発現の報告を見ると、宿主/ベクター系はX-33/pPIC Zαが多数派だと感じる。本記事で紹介するのはGS115/pPIC 9なので留意されたいが、多数派の方を考慮し、適宜X-33/pPIC Zαの場合について注釈・併記をしたい(その場合は*で示す)。
ピキア株を販売しているのは実質Invitrogen社のみ(現在はThermo Fisher Scientific社のグループ)である。それから、ピキアによる発現の手引きは成書がある (3) ほか、Thermo Fisher Scientific社のHPからマニュアルpdfがダウンロードできる (4,5) のでそちらを参考にされたい。
以下、ジャーファーメンターを使用する場合の操作法について述べる。
装置・器具・試薬
- ジャーファーメンター(各社):ただし、pH電極、溶存酸素(DO)電極を備えたもの(図1)(DO電極:培地中の溶存酸素量をモニターするための電極)
- pPIC9プラスミド(もしくは*pPIC Zαプラスミド)。あらかじめ目的タンパク質のcDNAを組み換えたものを作製しておく。
- Pichia pastoris GS115株(*pPIC Zα使用の場合はX—33株) (上記のP. pastoris GS115株、pPIC9プラスミドはThermo Fisher Scientific社の「Pichia Expression Kit, original kit(カタログ番号K171001)」に、X-33株とpPIC Zαプラスミドは同社の「EasySelect™ Pichia Expression Kit(カタログ番号K174001)」に含まれる)
- 一般的な培養装置類(各社)
- オートクレーブ
- 30℃恒温槽(エアインキュベーター)
- 試験管用の恒温振盪培養器
- バッフルフラスコ、500 mL 1個
- 500 mLバッフルフラスコ用の恒温振盪培養器
- エレクトロポレーション用装置
- 遠心機
実験スケジュール概要
大腸菌では培養の都度、コンピテントセルの形質転換を行うが、ピキアの場合一度形質転換すれば、その株をグリセロールストック保存できるので、培養の都度形質転換を行う必要がない。そこで、ピキアの形質転換部分と本培養の部分の手順を分けて述べる。
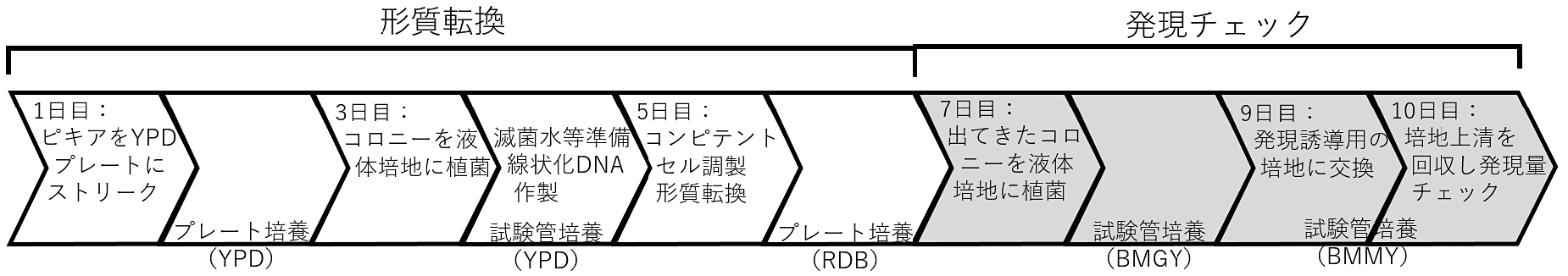
形質転換と発現チェック(スキーム1)
- 第1日
- ピキア株をグリセロールストックからYPDプレートにストリークし30℃でインキュベート。コロニーが現れるまで2~3日かかる。
- 第3日
- コロニーを液体YPD培地に植菌し、試験管で振盪培養(150rpm程度)する。2日間。
- 第5日
- 滅菌水やソルビトール溶液を用いてコンピテントセル液を調製する。そのコンピテントセル液にプラスミドを混合し、エレクトロポレーションで形質転換する。エレクトロポレーション後、菌体をセレクション用のプレートにスプレッドし、30℃でインキュベートする。2日間。
- 第7日
- コロニーが出てきたらそれぞれのコロニーを試験管に分注した液体培地(BMGY)に植菌する。30℃で2日間振盪培養(150rpm程度)。
- 第9日
- 菌体が増えたら培養液を遠心し上清を捨てる。その後、発現誘導用の培地(BMMY)で再懸濁し、再び振盪培養する。
- 第10日
- 培養液を遠心し、上清をSDS-PAGEなどで調べ、タンパク質が発現しているかどうかチェックする。
スキーム1. 形質転換と発現チェック
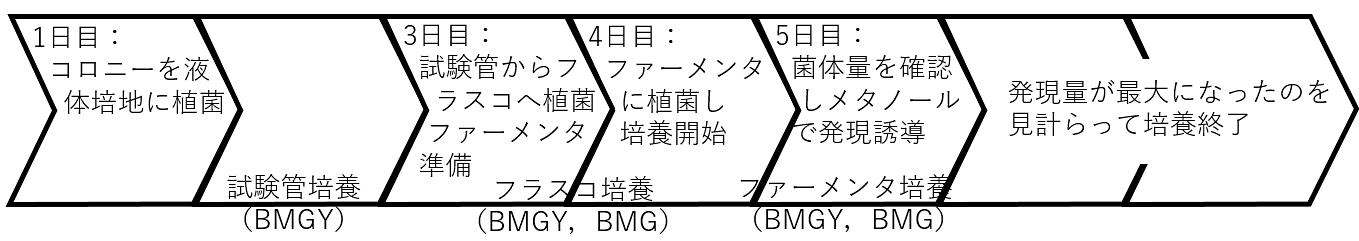
本培養(スキーム2)
- 第1日
- 発現株のコロニーをBMGY培地に植菌し、試験管で30℃で振盪培養する。2日間。
- 第3日
- 試験管の培養液をバッフルフラスコに植え継ぎ、引き続き旋回培養(120rpm程度)する。ファーメンターのセットアップを行う。
- 第4日
- 培養液をバッフルフラスコからファーメンターに植え継ぎ、ファーメンター培養開始。
- 第5日
- 菌体が一定量に増えたらメタノールを添加し、発現誘導する。
- 第6~8日
- タンパク質の発現状況をモニターし、発現が最大になった時点を見計らってファーメンターを止める。培養液を遠心し、上清からタンパク質の精製を開始する。
スキーム2. 本培養
実験の詳細
形質転換と発現チェック
第1日
ピキアGS115株(*X-33株)をグリセロールストックやスラントからYPDプレートにストリークし30℃でインキュベート。2~3日経つとコロニーが現れ1 mm弱の大きさに育つ。なお、このコロニーの生えたYPDプレートは2ヶ月くらいは冷蔵保存できるので、その間はストリークからの作業を行う必要はない。
第3日
コロニーを24 mLの液体YPD培地(含20 μg/mlクロラムフェニコール)に植菌し、試験管6本に分注し振盪培養(150rpm程度)する。2日間経つと菌体が十分に増える。この振盪培養期間中に以下の準備をしておく。
- 1 Mソルビトール溶液を100 mL調製しオートクレーブ滅菌した後、冷蔵庫で冷やしておく。
- 同様にイオン交換水を100 mLオートクレーブ滅菌した後、冷蔵庫で冷やしておく。
- 目的のタンパク質のcDNAを組み込んだプラスミドを適当な制限酵素(pPIC9ならStu IやBgl II、pPIC ZαならSac I, Pme I, BstX Iなど。どの酵素を用いるかは工夫とコツ『今回説明したピキア株‐プラスミド系について』参照)で線状化し、フェノクロ、エタ沈で精製、乾燥した後、滅菌水で溶解し、0.3~1.2 μg/μL(結構濃いめ、ミニプレップ後の5~10倍程度)にしておく。
第5日
以下の手順に従って、培養液から余分な培地を取り除き、菌体のみを集め、それを再懸濁することでコンピテントセル液を調製する。なお、温度が上昇すると形質転換効率が落ちるため、エレクトロポレーションまでの操作は氷上で行う。
- 振盪培養後の培養液を15 mLのコーニング管に移し、4500 g、3分で遠心する。遠心後培地上清は捨てる。
- 集められた菌体に10 mLの冷滅菌水を加え注意深く菌体を再懸濁し、遠心後、上清を捨てる。これを3回繰り返す。
- 集められた菌体に10 mLの冷1 Mソルビトールを加え注意深く菌体を再懸濁し、遠心後、上清を捨てる。これを2回繰り返す。
- 集められた菌体に適当量の冷滅菌水を加え、菌体懸濁液を適当な濃度にする。適当な濃度とは、できるだけ濃くはあるが、菌体の塊はなくマイクロピペットで引っかからずにゆっくり吸い上げられるくらいを目安とする。
その後、コンピテントセル液とプラスミドを混合しエレクトロポレーションで形質転換。
- コンピテントセル懸濁液40 μLに線状化プラスミドDNA液1~3 μLを混ぜる。
- エレクトロポレーション用のキュベットに入れ、1.7 kVで一回パルスを印加する。(条件は機器と電極間距離によって異なる。上記はBIO-RAD社製E.coli pulserと0.2 cmキュベットの場合の例。)
- 1 Mソルビトール溶液で菌体を回収し、菌体をHis無しのRDB寒天培地(*Zeocinを含むYPD寒天培地)にスプレッドする。30℃で2日間インキュベートする。
インキュベートの結果得られたコロニーは形質転換がうまくいった株である。しかし株によって発現量が異なるため、続く第7日目以降はスモールスケールで発現チェックを行う。
第7日
得られたコロニー(図2A)のうち10個ほど選び番号を振る。各コロニーを試験管に2 mLずつ分注したBMGY培地に植菌する。同時にそれらのコロニーを番号が書かかれたRDBプレート(図2B)にも植える。30℃で2日間、試験管は振盪培養、プレートは静置インキュベートする。
第9日
菌体が増えたら試験管内の培養液を2 mLのエッペンドルフチューブに移し、遠心して上清を捨てる。その後BMMY培地500 μLで再懸濁し、元の試験管に戻した後、振盪培養を再開する。
第10日
再びエッペンチューブに培養液を移して遠心し、上清を分取する。上清中の発現タンパク質の量をSDS-PAGEやブロッティングなどで調べ、どのコロニー株がより多くのタンパク質を発現するかチェックする。
ここで発現量の最大だった株を、以降の本培養で用いる。プレート上のピキアは2か月程度であれば、冷蔵保存可能で、その間は本培養に用いることができる。しかし、長期の保存用として、形質転換株をグリセロールストックにして-80℃で保存しておくのがよい。(グリセロールストックの作製法は工夫とコツ『グリセロールストック作製法』参照。)
実験の詳細 本培養
第1日
発現チェックで選択した株のコロニーを10 mLのBMGY培地に植菌する。その培地を5本の試験管に分注し、30℃で2日間振盪培養(150rpm程度)する。
第3日
試験管の培養液を100 mLの培地(BMGY or BMG)が入ったバッフルフラスコに植え継ぎ、引き続き旋回培養(120rpm程度)。
加えて、ファーメンターのセットアップを行う。
- pH電極の較正:通常のpH電極と同様、pH標準液を用いて2点較正する。
- DO電極の較正:大気較正と標準液較正の二つの方法がある。
- 大気較正:飽和点のみの1点で較正する。飽和点はDO電極の先端を空気中にさらししばらく置いた時の読み値とする。
- 標準液較正:酸素濃度0点と飽和点の2点で較正する。各点の溶液は以下のようにして調製する。
- 0点:四ホウ酸ナトリウム(25 g/L)と亜硫酸ナトリウム(100 g/L)の混合水溶液
- 飽和点:ファーメンターに脱イオン水を入れ、本培養時と同じ温度、攪拌速度、送気量にしてしばらく置いたもの。
- 培地1 Lの調製(BMGY or BMG)
- ファーメンターのオートクレーブ滅菌
第4日(グロース期)
培養液をバッフルフラスコからファーメンターに植え継ぎ、ファーメンター培養開始。はじめは菌体量が低いのでDO値は高い値を示すが、菌体量の増加に伴いDO値は減少する。培養を続けているとDO値が上昇する。その時にはグリセリンを補給する。
第5日(発現誘導期)
菌体が湿重量で150 g/L以上に増えたら培地をBMMY(合成培地の場合はBMM)に交換するか、メタノールを添加し、発現誘導する。(もっと菌体を増やしてから発現誘導をかけると発現量が増える場合もある。)
グロース期と同様、DO値が上昇したらメタノールを補給する。(もしくはポンプで一定速度の添加で、発現量が上がるとの報告もある。(6))
第6~8日
タンパク質の発現状況を電気泳動やHPLCでモニターし、タンパク質の発現が最大になった時点を見計らってファーメンターを止める。何度か培養を行い、ある程度発現量の頭打ちのタイミングがわかってきたら、時間を決めて培養終了しても良い。
培養終了後に培養液を遠心し、上清からタンパク質の精製を開始する。
工夫とコツ
DO電極使用の目的は?
DO電極は培養液中の溶存酸素量を計測しているものである。培養液中の溶存酸素量は培養液中に存在する炭素源の量と関係がある。つまり、炭素源が充分量あるとピキアの好気呼吸のため培養液中の酸素が消費され、溶存酸素量は減る。しかし、炭素源が枯渇すると酸素が消費されなくなるので、培養液中の酸素濃度が上昇する。つまり、DO値から培養液中の炭素源が枯渇していないかどうかが分かるのである。
今回説明したピキア株‐プラスミド系について
そもそもピキアにプラスミドが取り込まれるとそのプラスミドはピキアのゲノムDNAに組み込まれる。(そのため、プラスミドを線状化しておく必要がある。)GS115株はヒスチジン合成酵素の遺伝子に機能欠損の変異が入っているため(his4)ヒスチジン要求性であるが、pPIC9が持つ正しい合成酵素の遺伝子(HIS4)が組み込まれることによって、ヒスチジン要求性がなくなり、これで形質転換株のセレクションが可能となる。
なお、ひとつのピキアに複数のプラスミドが組み込まれる場合がある。これをマルチコピーというが、そのような株の方が高発現量になることが多い。そこでマルチコピー株を選択できるようにpPIC9KやpPIC Zαといったプラスミドもデザインされている。これらのプラスミドには抗生物質耐性遺伝子が含まれており、pPIC9Kは形質転換株のセレクションマーカーがカナマイシン耐性となり、pPIC ZαはZeocin耐性となる。培地中の抗生物質濃度を変化させることでより多くのコピー数の株をセレクションできる。
筆者はあまりこれらのプラスミドの使用経験がないが、Thermo Fisher Scientific社はこのpPIC9KやpPIC Zαを用いた発現系を推奨しているようだ。(それぞれ商品名は「EasySelect™ Pichia Expression Kit(カタログ番号K174001)」(5)、「Multi-Copy Pichia Expression Kit(K175001)」(7)である。)詳細は各マニュアルを参照されたい。
今回紹介したGS115-pPIC9の系で触れておかなければならない点がもう一つある。それは、宿主‐ベクターの組み合わせが同じでも、プラスミドを線状化する時に用いる制限酵素によって、形質転換株の表現型がMut+になったりMutSになったりするということである。Mutとは「Methanol Utility」、つまりメタノール資化性のことであり、Mut+はメタノール資化性の表現型、MutSはメタノール資化性はあるものの代謝速度が遅いという表現型を示す。(MutSのSはSlowのSである。)使う制限酵素と得られる表現型は以下のように対応している。
| 用いる制限酵素 | 得られる表現型 |
|---|---|
| Stu I(Aat I), Sal I | Mut+ |
| Bgl II | MutS |
表現型が異なる理由は、用いる制限酵素によってプラスミドが組み込まれるゲノム上の位置が異なり、その中でBgl IIを用いたプラスミドが組み込まれる時のみ、AOX1という遺伝子の欠損が起こるからである。AOX1とはアルコールオキシダーゼをコードする遺伝子であり、この欠損によりメタノール代謝速度が遅くなる。だからMutS株の場合、誘導期の期間をMut+よりも長くとる必要がある。(この辺の詳しい機構についてはオンラインpdfマニュアル等参照 (4)。)
しかしながら、目的タンパク質によってはMut+よりMutSの方が高発現量になるので、発現チェックはMut+、MutS両方を試すことが推奨されている。本稿ではMut+株の場合を想定して培養時間等を述べている。
なおX-33/pPIC Zαの系では上で挙げたSac I, Pme I, BstX Iのいずれの制限酵素でプラスミドを線状化してもMut+が得られる。MutSを試したい場合は異なるピキア株(KM71H)を用いる必要がある (5)。
各培地の説明
一連の手順に様々な培地が登場したが、それぞれがどのような目的で用いられているか説明する。
- YPD(液体、プレート)
- イーストエキストラクト(Y)、ペプトン(P)、グルコース(D)からなる培地。単純にピキアの菌体量を増やしたい時に用いる。
- RDB(His不含)(プレート)
- ヒスチジンを含まない培地。形質転換したGS115株のみ増えるので、セレクション用として用いる。
- BMGY(液体)
- 発現が目的の培養に用いる。炭素源としてグリセリンを含んでおり、グロース期に用いる。
- BMMY(液体)
- 発現が目的の培養に用いる。炭素源としてメタノールを含んでおり、発現誘導期に用いる。組成は炭素源以外BMGYと同じ。
- BMG/BMM(液体)
- 様々な無機塩類と、唯一の炭素源としてグリセリン(BMG)もしくはメタノール(BMM)、唯一の窒素源として塩化アンモニウムを含む合成培地。NMR測定試料タンパク質を発現させるときにはこの培地を使用する。大腸菌のM9培地のようなもの。
これらの培地のレシピは上述のThermo Fisher Scientificのマニュアルpdfで見られるほか、筆者が研究室内向けに作成したpdfにもあるので、いずれかを参照されたい。
ちなみに、筆者は同位体標識をしない時でも合成培地であるBMGを用いる時がある。発現タンパク質によっては発現量が落ちず、その場合はむしろ培地中の成分が少ないので後の精製が楽だからである。しかも天然培地に比べ、ファーメンター培養時の匂いが少ない。
もちろん、天然培地の方が高発現量になるタンパク質もある。この場合は天然培地に含まれるペプトンが発現タンパク質の加水分解を軽減するからだと考えられる。
グリセロールストック作製法
形質転換後のピキア株は、試験管培養で菌体量を十分に増やした後、最終濃度15%のグリセロールを加えることで、冷凍保存しておくことができる。簡単に作製手順を述べる。
発現チェックで選択した株のコロニーを2 mLのBMGY培地もしくはYPD培地に植菌する。30℃で試験管振盪培養を2日間行う。 ↓ 菌体が十分に増えた後、滅菌済みの70%グリセリンを550 μL加えて混合し、複数のエッペンドルフチューブに200~500 μLずつ分注する。その後、液体窒素ですばやく凍結し、-80℃で冷凍保存する。
培地中に加えるメタノール量を多くし過ぎない
ピキアの発現系では培養液にメタノールを加えることで発現を誘導するわけだが、これは決してピキアがメタノールに対して耐性があるというわけではない。ピキアにとってもメタノールは毒である。だから培養液中のメタノールはあまり上げない方がよい。そして、培地中のメタノール濃度が一定に保たれている方が高発現量になる (4)。メタノールセンサーを用いて最適なメタノール濃度を保つという研究もあるが、簡便にはポンプなどで少しずつメタノールを添加し、培地中のメタノール濃度を常時0.5%程度に保つとよい。
電極の扱い
ピキアのファーメンター培養では専用の電極でpHとDOをモニターする必要がある。これらの電極は基本的にオートクレーブ可能である。しかし、電極の横に空いている穴をビニールテープなどで塞いだままオートクレーブしてしまうと電極内部が陰圧になってしまい、正しい値を示さなくなる。特にpH電極ではその陰圧のせいで中のガラスが割れてしまい故障につながる。電極内に培地が入ることをおそれず穴を開放したままオートクレーブするべきである。またDO電極の表示値が不安定になる原因として、先端のねじ止め部(液絡)に培地や菌体のごみがたまって導通が悪くなっているケースがある。
ピキアが凝集するのだが!?
いつもは培養液で菌体が均一に分散しているのに時々菌体が凝集しているのを見かける。しかし、目的タンパク質が発現しているのなら問題はない。この凝集はpHが3.5付近になると起こるもので、培養中の代謝物で培地が酸性化するためと考えられる。pHを戻せば再び均質になる。ピキアはpH 3~7で普通に生育し発現するが、pH 8以上ではすぐ死んでしまうので注意する。
NMR用同位体ラベルをするときの注意点
ピキアの培養では合成培地(BMG/BMM)を使った条件も確立されている。BMG/BMMの窒素源は塩化アンモニウム、炭素源はグロース期がグリセリン(BMG)、誘導期がメタノール(BMM)のみであるため、\(\ce{^{15}N}\)や\(\ce{^{13}C}\)リッチな試薬に置き換えれば異核共鳴実験用の試料が得られる (8)。
また、発現タンパク質の炭素はメタノール由来だから誘導用培地中のメタノールのみ\(\ce{^{13}C}\)標識されたものを用いればよいのでは、という指摘もあるが、誘導初期にはグロース用培地中の\(\ce{^{12}C}\)炭素源由来の細胞内代謝産物も用いられ、同位体標識されていないタンパク質が発現してしまう。そのためグロース期から\(\ce{^{13}C}\)試薬を用いる必要がある (9)。グロース期の炭素源はグルコースでもよく、グルコース枯渇後30~60分おいてからメタノール添加すれば発現は阻害されない。\(\ce{^{13}C}\)グルコースは\(\ce{^{13}C}\)グリセリンより安価なためコスト低減になる。
N末端の配列の違いがタンパク質の性質を変えてしまう場合がある
発現タンパク質のN末端に余分なアミノ酸が付加してしまう場合がある。分子ごとに加水分解位置が異なる場合もある。N末端が柔軟で構造を取らないタンパク質では問題にならない可能性があるが、N末端までしっかり構造を取るタンパク質では安定性が大きく変化したとの報告がある (10)。気になる場合は特定の加水分解配列を導入し、発現後酵素で切断するなどの対策を取る。また、プライマー設計、合成培地化、培養温度、培養中のpH調整等で付加を抑えられる場合もある。
精製は、できることならCM-Sepharoseがよい
培養液上清から発現タンパク質を精製するとき、もし可能ならCM-Sepharose(Cytiva)の使用を勧める。ピキア培養液上清の不要タンパク質や核酸はCM-Sepharoseに結合しないためである。ウシβラクトグロブリン(等電点4.6)ではCM-Sepharoseワンステップで十分な純度が得られた経験がある (1)。ただし陽イオン交換カラムのためpHを下げる必要があり、低pHで不安定なタンパク質では上清中の酸性プロテアーゼによる分解に注意が必要である。
糖鎖付加について
発現タンパク質にAsn-X-SerやAsn-X-Thrという配列があると、そのAsn側鎖に糖鎖付加が起こる場合がある。この場合ハイマンノース型糖鎖(GlcNAc 2つとMan 8個程度)とハイパーマンノース型糖鎖(GlcNAc 2つとMan数100個)のいずれかが付加され、それらが混在して発現する。どのような状態の発現タンパク質を得たいかによって、ゲルろ過でハイマンノース型とハイパーマンノース型を分離するか、糖鎖除去酵素(Endo H、α1-6 Mannosidase、α1-2,3,6 Mannosidaseなどがよく使われる)で糖鎖を取り除くか、対策が異なってくる。
なおハイパーマンノース型は酵母特有の糖鎖である。ハイマンノース型は哺乳類でも生じるが、哺乳類細胞では多くの場合そのハイマンノース型から種々のプロセスを経て異なる糖鎖型になる。よってピキアの発現系では哺乳類細胞と同じ糖鎖を付加させるのは難しい。しかし、ピキアの糖鎖付加関連の遺伝子を改変することで、哺乳類型に近い糖鎖付加をさせる系の研究もなされている (11)。
文献
- Sakurai, K. & Goto, Y., J. Biol. Chem., 277, 25735-25740 (2002)
- Kozhukh, G.V. et al., J. Biol. Chem., 277, 1310-1315 (2002)
- Pichia Protocols: Methods in Molecular Biology, 2nd edition (ed. Cregg, J. M.), Humana Press, NJ (2007)
- https://documents.thermofisher.com/TFS-Assets/LSG/manuals/pich_man.pdf
- https://documents.thermofisher.com/TFS-Assets/LSG/manuals/easyselect_man.pdf
- Zhang, W. et al., In: Pichia Protocols: Methods in Molecular Biology, 2nd edition (ed. Cregg, J. M.), Humana Press, NJ (2007). https://doi.org/10.1007/978-1-59745-456-8_4
- https://documents.thermofisher.com/TFS-Assets/LSG/manuals/pichmulti_man.pdf
- Wood, M. J. & Komives, E. A., J. Biomol. NMR, 13, 149-159 (1999)
- Rodriguez, E. & Krishna, N. R., J. Biochem., 130, 19-22 (2001)
- Goda, S. et al., Protein Eng., 13, 299-307 (2000)
- Vervecken, W. et al., In: Pichia Protocols: Methods in Molecular Biology, 2nd edition (ed. Cregg, J. M.), Humana Press, NJ (2007). https://doi.org/10.1007/978-1-59745-456-8_4
改訂履歴
2025年9月22日 改訂
- 著者所属の情報の更新
- 英文タイトルの追加
- ピキアX-33株、pPIC Zαプラスミドに関する記述を追加
- 糖鎖付加に関する記述を追加
- 文献の追加、リンクの修正
- 培地レシピを修正
その他、全体に渡って、語彙・文章を適宜修正・追記
改訂前の PDF-

図1:(A)上:pH電極、下:DO電極。いずれもMETTLER TOLEDO社製。オートクレーブ可能である。(B)ジャーファーメンター。三ツワ理化学社製(現社名三ツワフロンテック)。ジャーの上部にpH,DO電極が取り付けられているのが見える。 -

図2:(A)形質転換の結果得られたピキアのコロニー。(B)プレートにマス目を描き、発現チェックする株を番号ごとに植えたもの。
