

概要
一言でX線溶液散乱測定といっても、その対象は多岐にわたるが、ここでは特に、蛋白質溶液からのX線散乱について述べる。図1に示すように、蛋白質溶液からのX線散乱は、散乱角に依存して、ほぼ単調に減少していく曲線を描く。結晶格子散乱の場合と同様に、散乱角が大きくなるにつれて、散乱曲線には、より高分解能の構造情報を含まれるようになる。通常、X線溶液散乱といった場合には、おおよそ分解能にして15Å程度(散乱角2.5~4度)までの散乱を用い解析する。比較的強度の強い小角領域の散乱(X線小角散乱)からは、蛋白質のサイズ、分子量等の構造情報を抽出することができる。これらの情報は、ゲル濾過クロマトグラフィーから得られる情報と相補的であるが、様々な溶液条件で、これらの物理量を直接測定できるという点のみを考えても、X線溶液散乱測定の優位性は十分理解できる。一つの検体あたり、一連の測定に要する時間は、数分から数10分程度であり、測定時間だけを考えれば、ラボで標準的に行う分光学的測定と同程度である。また、必要とする試料も、大きめのセルでUVスペクトル測定をした場合に使う程度の量(~数100μg)ですませることも可能である。更に、近年では、解析技術が進み、一見、何の変哲もない減衰曲線から、直感的にイメージしやすい実空間像(表面形状)を予測できるようになった。この結果、以前に比べ、専門的な知識がない研究者でも容易にX線溶液散乱曲線を解釈できるようになっている。本稿では、これらの解析に耐えうる、”信頼性の高い”X線溶液散乱曲線を測定するための方法と、具体的なアプローチについて説明する。
装置・器具・試薬
- X線溶液散乱用セル(自作、もしくは共同研究者のセルを借りる必要有り;図2)
- X線(KEK/PF製、SPring-8製、X線発生装置)
- Windows XPが動作する程度のPC
- 解析ソフト(IgorやKleidaGraphがあれば、大抵のことはできる)
- Svergunのソフトウェア群(http://www.embl-hamburg.de/ExternalInfo/Research/Sax/index.html)
実験手順
実験企画段階
1)放射光利用申請
ビームタイムまでの期間
2)予備実験(ゲル濾過クロマトグラフィー)
ビームタイム直前
3)サンプル調製(濃度定量)
ビームタイム当日
4)放射光利用実験(X線溶液散乱測定、予備&本実験)
ビームタイム後
5)事後確認実験(濃度定量、SDS-PAGE)
6)解析
実験の詳細
実験企画段階
1)放射光利用申請
他の測定手法同様、X線溶液散乱測定を行うためには、それ相応の装置が必要である。自前でそろえるとなると6千万円程度の予算を必要とするため、少し測ってみたいという研究者には敷居が高い。しかしながら、幸い、X線溶液散乱実験を行える共同利用研究機関が、国内に複数箇所有り、課題申請をすることで、公的研究/教育機関に所属している研究者であれば、誰でも実験することができる。施設により異なるが課題公募は年2回程度の頻度で行われている。
- 高エネルギー加速器研究機構 放射光科学研究施設 Photon Factory
主なビームライン:BL-10C, BL-15A
申請に関する詳しい情報:http://pfwww.kek.jp/users_info/summary/indexj.html - 大型放射光施設 SPring-8
主なビームライン:BL40B2, BL40XU, BL45XU
申請に関する詳しい情報:http://www.spring8.or.jp/ja/support/ - その他(工夫とコツ参照)
とはいえ、やはり初めて実験する人は、学会や論文などでX線溶液散乱測定の結果を発表している人を捕まえ、共同研究という形で申請し、実験する方が安心だろう。
ビームタイムまでの期間
2)予備実験
課題が採択されても、実際のビームタイムまでは、はやくても数ヶ月待たなくてはいけない。この期間を利用して、もし、試料が許すのであれば、予備実験をしておくことが望ましい。特に、蛋白質の氏素性をそれなりに理解しておくことは重要である。蛋白質の溶液中での存在状態が単分散であるのか、多分散であるのかによって、実験の仕方や適応できる解析法、実験結果の解釈の仕方が全く異なってくる。従って、実際に実験してみようと思っているのに近い溶液条件で、ゲル濾過クロマトグラフィーなどを行い、試料の会合性について把握しておく必要がある。特に、凝集体が少しでも観測される条件では、X線溶液散乱曲線の解釈が困難になるので、溶液条件の検討(塩濃度やpH)を行う必要がある。
ビームタイム直前
3)サンプル調製
X線溶液散乱測定の大敵は、蛋白質の非特異的な会合である。他の測定手法に比べ、X線溶液散乱測定は、会合体形成に極めて敏感であり、凝集体が微量存在するだけで、結果を解釈するのが難しくなる。従って、この非特異的な会合をいかにして避けられるかが、実験の正否の分かれ道となる。このような会合を避けるためにも、試料はできるだけフレッシュであることが望ましい。仮に、凍結乾燥ストックや冷凍ストックなどを使うのであれば、凍結融解等で生じた非特異的な会合体を、ゲル濾過クロマトグラフィーで取り除く等の一手間を加えると、結果が格段に改善される。
使用する装置に依存する部分もあるが、[重量濃度](mg/ml)×[分子量](kDa)が~45程度を満たす蛋白質濃度以上であれば、”容易に”測定できる。(これ以下は、測定できないという訳ではない。より低濃度の場合には、それなりに気を遣った測定をしなくてはいけない。) 信頼性の高いX線散乱曲線を得るためには、複数の異なる蛋白質濃度条件で、X線溶液散乱を測定する必要があり、目安から算出された濃度x1, 2, 3 …NumMaxの試料を用意しなければならない(著者等の場合、Nummaxは>6を目安にしている)。そこで、先にゲル濾過クロマトグラフィーによって精製された試料を、~45/[分子量](kDa)x NumMaxまで濃縮し、その後、緩衝液を用いて、目的の濃度に希釈する。濃縮には、一般的な限外濾過器を用いる。希釈に使う緩衝液は、ゲル濾過クロマトグラフィーに用いたもの、透析したのであれば透析外液、もしくは、限外濾過で生じる濾液などを使う。X線溶液散乱測定のセルは、著者等の場合、分注の簡便さも考慮して、容積20μl程度のものを使用している(図2参照)。従って、45kDaの蛋白質の場合、1, 2, 3, 4, 5, 6 mg/ml、各々25μlずつ用意したとして、約0.5mgの蛋白質があれば一連の測定ができる。むろん、ここに書いた条件は目安に過ぎず、濃縮が困難な試料については、濃縮可能な範囲内で実験しなくてはいけない。
ビームタイム当日
4)放射光利用実験(X線溶液散乱測定)
・X線のコリメーション
実験当日は、X線のコリメーションを行わなくてはならないが、誰でも最初からできるものではない。利用する施設によって運営が異なっているが、共同研究者や施設のビームライン担当者の協力が必要不可欠である。
・X線を用いた予備実験
いったん、X線のコリメーションが終わってしまえば、UVスペクトル測定と同様に、淡々と試料を入れ替え測定するだけである。しかし、新規の試料を測る際には、入念な予備実験が必要である。
放射光は、非常に強力な光源で、長時間試料に照射すると、試料にダメージを与える。ダメージの受けやすさは、蛋白質の素性によって異なり、一言で言える目安はない。従って、初めての試料を測定する際には、どの程度の露光時間であれば試料にダメージがないかを、あらかじめ検討する必要がある。著者等は、実際に測定しようとしている濃度系列の中から、中間濃度の試料を多めに作り、ダメージのチェックに用いるようにしている。同一試料に対し、繰り返しX線を照射し、その都度、散乱曲線を測定することで、照射時間依存的な散乱曲線の変化を確認する。試料がX線照射によってダメージを受けた場合、凝集体が形成される。凝集体形成は、小角領域に散乱の増加をもたらすので、これを指標として、同一試料に対する露光限界時間を把握する。また、同時に、解析に十分なS/Nを得るのに必要な全露光時間もこのときに確認する。仮に、全露光時間よりも、露光限界時間の方が短い場合には、逐次、試料を交換し、十分なS/Nが得られるよう散乱曲線を積算する必要がある。
著者等の経験があるビームラインについて、全露光時間の目安を以下に示す。
(ovalbumin,~45kDa, 2mg/ml)
| 施設 | BL | 検出器 | 波長 | 光路長 | 全露光時間 |
|---|---|---|---|---|---|
| KEK/PF | BL-10C | PSPC*1 | 1.5Å | 1mm | ~600sec |
| BL-15A | XII-CCD*2 | 1.5Å | 1mm | ~120sec | |
| SP8 | BL-40B2 | IP*3 | 1.0Å | 3mm | ~60sec |
*1RIGAKU製150mm1次元ガス検出器
*2HAMAMATSU製イメージ増感CCD型X線検出器
*3R-axis IV++
・試料の測定とバックグランドの測定
ダメージへの耐性、並びに、解析するのに十分なS/Nを得られる露光時間の見積もりが終われば、いよいよ本実験の開始である。蛋白質に由来する散乱曲線I(Q)=ΔIsamp(Q)の算出は、次式で表すことができる。
\[\Delta I_{samp}(Q) = \left( \frac{I_{samp}(Q) - I_{Dark}(Q)}{I_{0,samp}} \right) - (1 - \frac{c \nu}{1000}) \left( \frac{I_{buf}(Q) - I_{Dark}(Q)}{I_{0,buf}} \right), \hspace{5mm} Q = \frac{4 \pi sin \theta}{\lambda}\]一見煩雑な式であるが、蛋白質溶液のデータ(Isamp(Q))から緩衝液(透析外液、もしくは、限外濾過で生じる濾液)のデータ(Ibuf(Q)(Q))を引いているだけである。(正確には、空セルの散乱曲線も考慮する必要があるのだが、多くの場合、結果的に無視できるので省略した。)
ラボで一般に使われている分光光度計は、ダブルビームタイプのものが多く、入射強度の変動を気にしたことはないかもしれないが、X線では、ちょうどシングルビームタイプの分光光度計に近く、入射強度の変動を考慮しなくてはいけない。分子のI0,samp及びI0,bufは、それぞれ蛋白質溶液と緩衝液を測定している間の、入射X線積分強度である。IDark(Q)は、検出器に依存したもので、X線が入っていないときの装置固有のバックグラウンドである。cνは、それぞれ、蛋白質濃度(mg/ml)と蛋白質のPartial Specific Volume(偏比容;cm3/g)を表している。偏比容は、大雑把に言って1gあたりの蛋白質の体積(cm3)であり、(1- cν/1000)は、蛋白質溶液における、蛋白質以外の部分(溶媒)の体積分率に相当する。実測した蛋白質溶液と緩衝液の散乱曲線は、溶媒部分のみに注目すると、後者は前者に比べ、蛋白質の分だけ、より多く溶媒由来の散乱を含むことになる。そこで、上述の補正項を緩衝液の散乱曲線にかけたものを引く必要がある。偏比容は、蛋白質によって異なるのだが、おおよそ0.7~0.75cm3/gの値をとる。10mg/mlの蛋白質溶液の場合でも、この補正項は0.993~0.9925程度なので、部分比容は、例えばbovine serum albuminの0.735やlysozymeの0.712に固定しておいて問題ない。
まとめると、蛋白質の散乱曲線を求めるために必要なデータは、蛋白質溶液の散乱とその時の入射X線積分強度、緩衝液の散乱とその時の入射X線積分強度、及び蛋白質濃度である。試料の露光時間は、予備実験で得られた値に設定する。このとき、原則、蛋白質溶液と緩衝液の露光時間は同一にする。蛋白質溶液については、事前に用意した、複数の濃度条件について全て測定する。緩衝液は、蛋白質溶液の希釈に用いたもの(サンプル調製参照)を用い、一連の蛋白質溶液の測定の、最初、中程、最後の3回程度測定しておく。おそらく、ビームタイムでは、複数の蛋白質、複数の溶液条件で測定することが多いと考えられるが、基本は、上の繰り返しになる。
・標準試料の測定
装置の状態や、実験の再現性の確認、更には見かけの分子量を算出(後述)するために、標準試料の測定をビームタイム毎に行うようにする。著者等は、標準試料として、bovine serum albumin(~66kDa), ovalbumin(~45kDa), myoglobin(~17kDa), hen egg white lysozyme(~14kDa)等を用いる。対象とする試料に比較的近い分子量を持つ標準試料を選択すると、後の解析の信頼性が向上する。X線は、塩によって吸収されるので、塩濃度によって散乱強度が変わってくる。従って、必ず、評価したい蛋白質溶液と同じ組成の緩衝液に溶かした状態で測定しなくてはならない。
ビームタイム後
5)事後確認実験
ビームタイム終了後、直ちに、実際に測定した試料の濃度定量、及びSDS-PAGEをすることを推奨する。測定後の試料は、大なり小なりX線によるダメージを受けているので、試料の減少量や分解をUVスペクトルやSDS-PAGEで確認する。著者等は、測定直前にX線溶液散乱測定用セルに入れたままUVスペクトル測定を行っているので(工夫とコツ参照)、その値も考慮して、測定結果の信頼性を評価している。
6)解析
・Guinier解析
本稿では、個々の解析については記述しないが、X線溶液散乱曲線を解釈する上で、全ての基本となるGuinier解析についてだけは簡単に解説する。X線小角散乱は、分子サイズに極めて敏感である。小角領域では、散乱強度I(Q)は次式のように展開できる。
\[I(Q) = I(0) \cdot \exp \left\{-\frac{Rg^2 \cdot Q^2}{3} \right\} + o(4), \hspace{5mm} (Rg^2 = \frac{\int\limits_V \rho (r)r^{2}dV}{\int\limits_V \rho (r)dV})\]ここで、Rg は慣性半径、I(0)は原点散乱強度を示している。o(4)は高次項の寄与を示しており、主に分子形状に依存する。Q<1/Rg程度の小角領域では、高次項の寄与が無視できるようになり、いわゆるGuinier近似が成立する。Guinier近似が成立する領域では、散乱強度の対数ln(I(Q))がQ2に比例することから、Q2 vs ln(I(Q))プロット(Guinierプロット)の傾きから慣性半径Rgを、y切片から分子量に依存する原点散乱強度I(0)を求めることができる。I(0)は、モル濃度x(MW)2に比例する値なので、試料の重量濃度(モル濃度xMW)で割ると、その値はMWに比例する。従って、分子量既知の標準試料をコントロールにすることで、対象とする蛋白質溶液の見かけの分子量を計算することができる。
多くの場合、見かけの分子量を用いて、会合数を評価することになると思う。このとき、予備実験で行った、ゲル濾過クロマトグラフィーの結果を考慮すると解釈が容易になることもある。例えば、X線溶液散乱から会合数1.5が得られた場合、単量体と多量体の平衡状態を観測しているか、実験上の問題で2量体もしくは単量体が、1.5量体として観測されたかの、大別して2種類の解釈ができる。幸い、ゲル濾過クロマトグラフィーでは、これらの事象が区別できる場合もある。仮に、ゲル濾過クロマトグラフィー上で2本以上のピークが観測された場合(多分散系)には前者、1本のピークしか観測されなかった場合(単分散系)には後者の可能性が高い。しかしながら、単量体と多量体の速い平衡の場合、ゲル濾過クロマトグラフィーにおいても一本のピークとして検出されることがあることから、濃度依存性を調べるなど、慎重に解釈することが肝要である。
・ゼロ濃度外挿散乱曲線の構築
ここまで、複数の濃度条件で測定することの意義について記述してこなかった。これらのデータは、Guinier解析から算出された物理量のゼロ濃度外挿値を算出するため、さらには、以後の解析のために必要な、ゼロ濃度外挿散乱曲線算出のために用いる。X線溶液散乱曲線は、1つの蛋白質から生じる干渉性散乱と、蛋白質間の距離相関に由来した干渉性散乱の積で表現される。蛋白質”溶液”とはいえ、蛋白質濃度が有限な限り、大なり小なり、蛋白質同士の距離秩序が生じる。従って、この効果を取り除かない限り、正確な、蛋白質分子由来の構造情報を読み解くことができない。具体的な例を、図3に示した。極端な例を示しているが、濃度に依存して、小角領域の散乱強度がはっきりと変化していることがわかる。この例を見てわかるように、濃度1点だけで測定した場合、その散乱曲線から得られるRgやI(0)には、蛋白質間干渉の影響を受けている可能性を排除できない。一般的には、各濃度条件で算出されたRg2やI(0)/conc.等を、濃度に対してプロットし、直線近似によってゼロ濃度外挿することで、蛋白質間干渉の効果を排除する。ゼロ濃度外挿値の精度は、測定する濃度点の数と測定間隔に依存するので、どの濃度で何点測定するかが、重要になってくる。著者等の感覚では、Guinier解析結果の外挿であれば、(使えるデータが)等間隔に5点程度測定できれば十分と考えている。
ゼロ濃度外挿は、Guinier解析だけでなく、構造予測解析や、その骨格を担う距離分布関数を計算する上でも、非常に重要な意味を持つ。これらの解析では、Guinier解析と異なり、小角領域の散乱だけでなく、得られた散乱曲線全体を用いる。従って、より信頼性の高い計算をするためには、散乱曲線そのものをゼロ濃度外挿する必要がある(図3、赤線)。散乱曲線のゼロ濃度外挿の方法は様々考えられるが、著者等は、近年、特異値分解解析を適応し、濃度依存項となるV-スペクトルをゼロ濃度外挿し、その値とU-スペクトルからゼロ濃度外挿散乱曲線を計算するようにしている。
・以後の計算
概要で述べた、”信頼性の高い”X線散乱曲線を得るとは、非特異的会合の効果が含まれず、蛋白質間干渉効果をうまく取り除けた、正しいゼロ濃度外挿散乱曲線を得るということである。ここまでに記した手順をふめば、ある程度”信頼性の高い”散乱曲線を求めることができ、少なくとも、どこに問題があるかは理解できていると思う。いったん”信頼性の高い”X線散乱曲線を求めてしまえば、Svergun等によって、公開されているプログラム群を利用し、後の解析を行うことができる。その中で、全ての解析の基本となるのが、GNOMという、半ばデファクトスタンダードとなったプログラムパッケージを使った距離分布関数の計算である。この計算には、ある程度慣れが必要だが、本稿ではその解説を省略する。一端、GNOMの解析が終われば、同じくSvergun等が公開しているプログラム群にGNOMのOutputファイルを流し込むだけで形状予測は行える。
工夫とコツ
濃度定量
上述のように、X線溶液散乱曲線を解析する時には、蛋白質濃度が特に重要な意味を持ってくる。従って、X線溶液散乱測定においては、1にも2にも濃度定量、3,4で散乱を測定し、5で濃度定量、といった意識が重要である。著者等は、後々後悔しないように、試料調整の際、全ての元になる最高濃度試料の濃度定量には細心の注意を払い、測定条件でのUVスペクトルの測定と変性剤存在下でのUVスペクトルの測定を、複数回実施し、濃度の再現を取るようつとめている。更に、分注後の蛋白質濃度を正確に測定するために、X線溶液散乱セルに入れた状態でUVスペクトルを測定することができる分光器を自作し、X線溶液散乱測定直前に、個別に、その場でUVスペクトルを測定するようにしている。更に、実験後、研究室に戻ってから、ストックの濃度を再度定量し、実験期間内に沈殿などによる濃度変化がないかを確認している。一連の実験を通じ、蛋白質を使った実験で、もっとも難しい実験は濃度定量であるという考えにたどり着く。
標準試料(原点散乱強度校正用)
どんな測定でもそうだが、標準試料の選び方は難しい。一長一短で、決定的なものはない。そんな中で、著者等が好んで使っているのが、ovalbuminである。分子量が45kDaと手頃な大きさであることもさることながら、会合しにくく、再現が非常に取りやすい。bovine serum albuminやhen egg white lysozymeも、場合によっては使うのだが、bovine serum albuminは2量体を形成しやすく、室温で放置したりすると、すぐに大きめのRg(慣性半径)、I(0)(原点散乱強度)を示す。lysozymeは比較的安定なのだが、バッファーを選ぶらしく、塩濃度や緩衝液の種類によっては、X線照射時に会合しやすくなって困る。それに対し、ovalbuminは緩衝液や塩濃度に対して、許容範囲が広く、非常に再現が取りやすい。酸性条件下でも、変性せず、標準試料として使える。酸変性状態を測定した人で、原点散乱強度が激減して解釈に困ったことはないだろうか?一度、ovalbuminを測定することをおすすめする。問題の解決はできないが、参考にはなる。
標準試料(散乱角校正用)
カメラ長、X線の波長と検出器の1chあたりの長さがわかれば、原理的には散乱角を求めることができる。しかし、実際には、正確にカメラ長や検出器の1chの長さを知ることは難しい。そこで、検出器のchと散乱角を対応づけるために、周期長既知の標準試料からの回折像を利用する。X線溶液散乱で、一般的に用いられるのはコラーゲンである。コラーゲンの周期長は、乾燥状態で653Åであり、X線溶液散乱を測定する領域に、明瞭な、多数の回折ピークを示す。しかし、さすがに生ものだけに、X線照射に伴って、周期長が変化し、知らない間に散乱角の校正がずれてしまうという、しゃれにもならないことが起こりえる。著者等は、養鶏場に就職したOBから、鶏の足を分けてもらい乾燥コラーゲンを調製している。鶏の足は想像以上に大きく、はさみを入れるのに勇気がいるが、腱はすぐに見つかり、1本の足から、使い切れないほどのコラーゲンを採取することができる。適当な長さに裁断し、コニカルチューブのそこで、軽く筋に沿ってこすり、ある程度乾いたところで、両端をダブルクリップではさみ、割り箸に通して吊しておく。この際、テンションをかけ過ぎると周期長が変わってしまうので、ダブルクリップの自重程度で十分である。日陰で干し、ねじれが生じてきたら、こまめに戻すようにして、最終的に、ねじれが生じなくなるところまで乾かしたところで、窒素パージしたコニカルチューブに保管する。校正に使うときには、常に、使い始めた時期が異なる、新旧2つのコラーゲンを測定する。両者のピーク位置にずれが生じ始めたら、古いものを捨て、新しいものをおろすようにする。この手順を踏むことで、散乱角の校正ミスは、防ぐことができる。
著者等は、この他にベヘン酸銀も測るようにしている。ベヘン酸銀は58.53Åの周期長を示す回折ピークを示すので、X線溶液散乱で用いる領域に、かろうじて1~2本程度のピークを観測することができる。回折ピークの本数が少ないので、校正用としては不便だが、コラーゲンのデータと相互比較することで、校正エラーが生じないようにしている。
ベヘン酸銀の購入先:
国内でも、バルクで購入することは可能である。しかし、下記のメーカーから購入すると、データテーブルも添付されてくるので、校正用として使う際にはおすすめである。著者等は、和研薬を通じて購入している。
Silver Behenate http://www.thegemdugout.com/products.html
Blanton T. N. et al. (1995) “JCPDS-International centre for diffraction data round robin study of silver behenate. A possible low-angle x-ray diffraction calibration standard” Powder diffraction 10(2), 91-95.
発生源の使い分け
通常、X線溶液散乱実験は、KEK/PFかSPring-8に設置されたビームラインを用いて行う。初めて実施するに当たり、困るのはどのビームラインを使えばいいかという点だと思う。結論から言えば、ここに示した程度の実験を行うのであれば、どこのビームラインを使っても実験できる。従って、余り悩まずに、出張旅費のかからない(近くの)施設を使って実験をするというスタンスでいいと思う。ただ、著者等の感覚では、腰を据えてゆっくりしたいテーマについてはKEK/PF、精度を要求する実験は気合いを入れて、SPring-8で行うようにしている。これは、光の性質が違うこともあるが、むしろ、申請実験の採択期間が、異なることが理由である。(KEK/PF 2年、Spring-8 半年(通常申請))
ラボではできない実験なのか?
決してそんなことはない。最近のX線発生装置は以前に比べ安定で、フラッグシップモデルを使わなくても、そこそこのフラックスが得られる。更に、Confocalミラーを使えば、従来の5倍程度のX線を集めてくることができる。検出器に関しても、従来であれば、1次元のガスカウンターや2次元のImaging Plateしか選択肢がなかったが、現在では、イメージ増感CCD型X線検出器(XII-CCD, HAMAMATSU)やPixel Array Detector等も商品化され、検出ロスが少ない検出器が購入できるようになった。著者等の研究室では、理学の少し前の回転陽極X線発生装置(UltraX18)、小角用Confocalミラー、XII-CCDの組み合わせで実験を行っている。光源強度は放射光と桁違いに弱いが(>2桁落ち?)、結果的には重量濃度x分子量~45の法則に従い、この条件を満たす試料であれば、1検体あたり20分程度で図4に示すようなデータを測定することができる。発生装置がかなり高いが、他の部分は、それほど非常識な値段ではない。すでに、結晶用としてX線発生装置を持っている研究室であれば、空きポートを活用することで、CDを2台買うぐらいの感覚で導入できるのではないだろうか?
ちなみに、上述のX線発生装置を用いた溶液散乱装置は京都・先端ナノテクノロジー総合支援ネットワークに登録されており、簡単な申請で使っていただくことができる(http://mswebs.naist.jp/nanotech/index.html)。本格的に使うのには躊躇するが、一度測ってみたい方は、是非申請していただきたい。
測定しやすいサンプル?しにくいサンプル?
測定しにくい、というか測定できないサンプルは、会合しやすいサンプルである。X線溶液散乱は、会合体形成に敏感なため、少量の非特異的な会合体が含まれている時点で、解釈が困難になる。仮に、生理的条件下で会合していなくても、X線照射によるダメージですぐに会合体を作ってしまうような試料もお手上げである。余り生産的なコメントではないが、著者等の経験から、結晶化したサンプルは、理想的なX線溶液散乱曲線を測定することができ、また、逆にX線溶液散乱測定が困難な試料は結晶化しない、という印象を持っている。結局のところ、非特異的な会合を示しやすい試料は、結晶もできなければ、溶液散乱測定もできない、つまり構造解析には向かないということなのだろう。いくら頑張っても結晶ができない試料については、占うつもりで、一度、溶液のままX線を照射してみるというのも手かもしれない。素直に測れたら、きっと・・・・!?
ダメージからいかに逃れるか?
会合しやすい試料は、X線照射損傷をきっかけにして、あっという間に会合が促進され、凝集体を形成する。X線照射損傷による凝集体形成を防ぐためには、蛋白質ができるだけ会合しない条件を探す必要がある。とはいえ、できることは限られており、せいぜい、塩濃度やpHを変える、グリセリンを入れる、等々、である。が、これで劇的に改善されることはほとんどない。そもそも、X線照射損傷の初期過程は何なのか?X線照射損傷の研究はディープなので、あまりコメントしない方がいいのかもしれないが、初期に形成されるラディカルが引き金になるのだろう・・、程度は言って差し支えないと思う。実際、著者等は、X線照射損傷がひどい試料には、DTTやメルカプトエタノール等の還元剤を添加するが、これが劇的にきく。とはいっても、それでも会合しやすいものは大なり小なり凝集体を形成してしまうので、結局お蔵入りになることも多いのだが・・・。
文献
X線小角散乱の教科書
- “Structure Analysis by Small-Angle X-Ray and Neutron Scattering”, L. A. Feigin and D. I. Svergun:生体物質に関するX線小角散乱の詳しい教科書。最近、広く普及しているビーズモデルを使ったAb initio形状予測についての話はあまり記述されていないが、球面調和関数を用いた形状”解析”は勉強になる。
- “Small-Angle X-Ray Scattering”, O. Glatter and O. Kratky (1982) Academic Press, London; “Small-Angle Scattering of X-Rays”, A. Guinier and G. Fournet (1955) Wiely, New York:いわずとしれた、X線小角散乱の名著。持っていなくても、参考文献に引いたことがあるのではないだろうか?General theoryはA. Guinier and G. Fournetの方が、応用面はO. Glatter and O. Kratkyの方が詳しい。
散乱・回折の教科書
Diffraction of X-Rays by Chain Molecules, B. K. Vainstein (1966) Elsevier, Amsterdam:X線散乱・回折を専門にしたいのであれば必読。
日本語の解説
- 意外に多い小角散乱実験からの情報(1)~(5) 結晶学会誌(1999), 41&42
これが全てだと思うのは危険だが、とてもよくまとまっているので、初めて実験をする人は目を通して損はない。図書館のアーカイブなどを検索してみよう。 - 実験化学講座10 回折 7章 丸善
X線溶液散乱の解説書ではないが、X線小角散乱について、比較的詳しく解説してある - X線の回折 三宅静雄 朝倉書店
X線回折一般の解説書だが、まじめにX線をやるなら一通り目を通した方がいい。
Svergunの論文
実際のところ、手っ取り早くX線溶液散乱を使った構造解析をしてみたいのであれば、Svergun等のHP(http://www.embl-hamburg.de/ExternalInfo/Research/Sax/index.html)を、一通り目を通すのが一番だと思う。“信頼性の高い”X線散乱曲線が測定できていれば、Default設定で、それなりにもっともらしい結果を出してくれる。中でも、In-direct法による距離分布関数計算(GNOM)に関する論文と、結晶構造からのX線散乱曲線計算(CRYSOL)に関する論文は”必ず”目を通すべきである。
- GNOM
“Determination of the regularization parameter in indirect-transform methods using perceptual criteria” D. I. Svergun, J. Appl. Cryst. (1992). 25, 495-503 - CRYSOL
“CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules fromAtomic Coordinates” D. Svergun, C. Barberato and M. H. J. Koch, J. Appl. Cryst. (1995). 28, 768-773
ここで報告されている解析法は、彼らが提案する全ての解析法の中核をなす。また、X線溶液散乱を理解する上で非常に役に立つ。特に、GNOMに関する論文は、彼らの蛋白質観知る上で非常に重要であり、GNOMを使う上での注意点がどこにあるのか理解することができる。Total estimate なるものが、彼らの蛋白質観の指標に過ぎないことが理解できると思う。(多くの場合、それが的を射ているところがすごいことなのだが。)
-
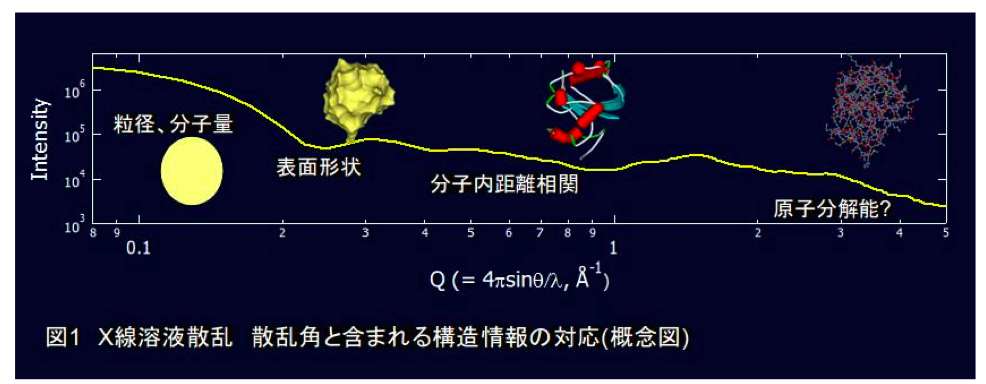
図1: -
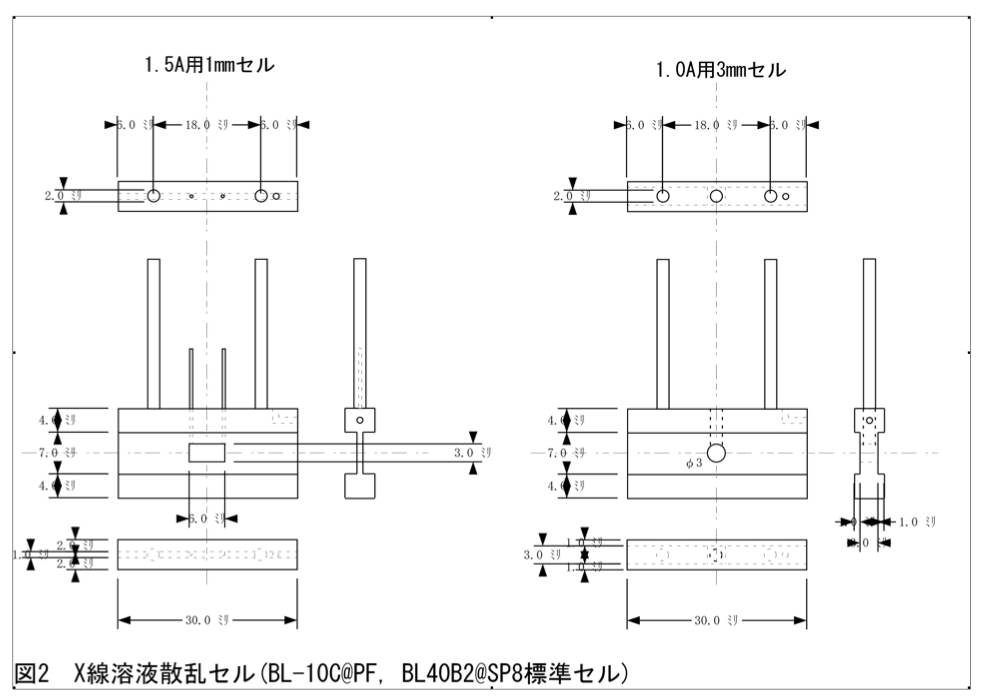
図2: -
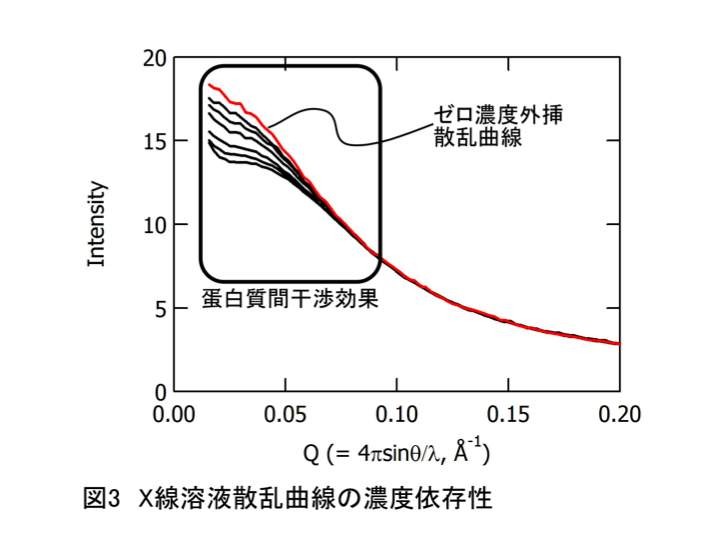
図3: -
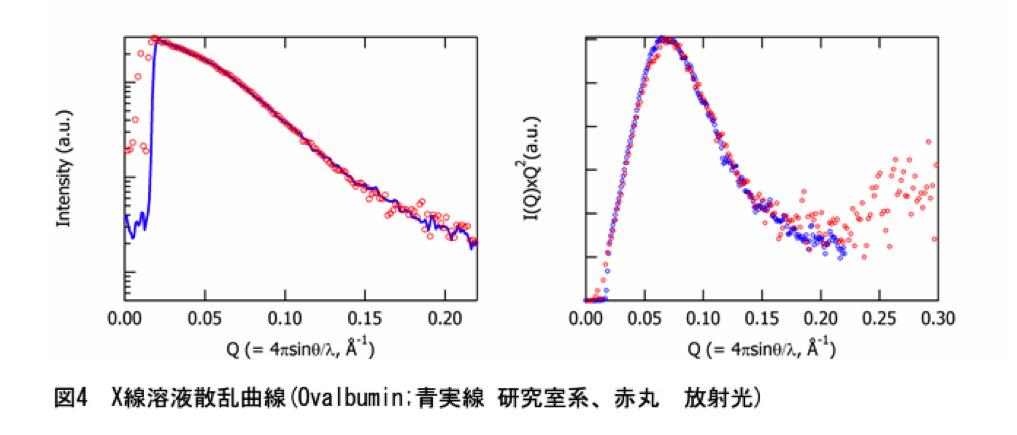
図4:
