

概要
蛋白質に結合する低分子化合物(以下、低分子リガンド)は、創薬標的蛋白質に対する分子標的薬や機能未知の蛋白質に対するケミカルノックダウンの手法としての応用が期待される。蛋白質と低分子リガンドの相互作用を検出する手法には本プロトコールで取り上げる表面プラズモン共鳴(SPR)1 の他に、等温滴定型熱量測定(ITC)、示差走査蛍光測定(DSF,もしくはいわゆる Thermal Shift Assay)、核磁気共鳴分光法(NMR)、マイクロスケール熱泳動(MST)、バイオレイヤー干渉法(BLI)など様々な物理化学的手法の選択が可能である2。しかしながら、低分子リガンドを大量の分子種からなる化合物ライブラリーの中からハイスループットに選抜しようとした際、多くの手法は使用する蛋白質の量・検出感度・スループット性の問題から必ずしも最適とは言えない。それに対し、SPR は比較的スループット性を担保した(本プロトコールで紹介する手法では、384ウェルプレート1枚あたりおよそ6時間)装置を用いることにより、酵素・蛋白質–蛋白質間相互作用に関わる蛋白質・膜蛋白質といった様々な標的に対する低分子リガンドを選抜することができる3–6。SPR の相互作用検出原理は表面プラズモン共鳴を活用した質量変化であるため、蛋白質に対する低分子リガンドの結合を、エンタルピー駆動・エントロピー駆動等の相互作用様式によらず選抜することも可能である。また、SPR のセンサグラムからは蛋白質-リガンド相互作用における速度論パラメータを議論することも可能であるため、これらの情報を活用することで一連の研究プロセスの早い段階にて擬陽性の化合物を排除することも可能である。したがって SPR による低分子リガンド選抜は、有効な低分子スクリーニング技術の1つとして挙げられる。
イントロダクション
SPR による相互作用測定ではセンサーチップ上に標的となる蛋白質を固定化し、そこへ化合物を一定の流速で流してゆく。蛋白質の固定化には様々な手法が用いられているが、本プロトコールでは Sensor Chip SA に対してビオチン化した蛋白質の固定化法について記す。この手法が選択された背景には、低分子と蛋白質の相互作用を測定するのに「安定的なベースライン」と「十分な蛋白質固定化量」が欠かせないことが挙げられる。前者に関しては、例えば Sensor Chip Ni-NTA に対する His キャプチャーを用いた固定化法では、実験中にセンサーチップ上から蛋白質が乖離し正確な結合測定が行えない問題がしばしば散見される。後者に関しては、例えば Sensor Chip CM5, CM7 に対するアミンカップリング法では、固定化に際し蛋白質を酸性 pH にさらすことや、蛋白質が多点で固定化されることに由来する蛋白質の失活が重大な固定化量の低下を招くことがある。こうした問題を同時に解消する可能性があるのが、本プロトコールで採用した SA-biotin キャプチャー法である。Streptavidin とビオチンの高い親和性から、実験中の蛋白質のセンサーチップ上からの解離はほとんど起こらない。さらに、ビオチン化修飾は蛋白質の N 末端か C 末端を選択して Avi-tag を付加し、タグに対しビオチン1分子が特異的に修飾される酵素反応を使用する7–9ため多点での蛋白質の固定化も防ぐことができる。そのため蛋白質の活性を保った状態で十分な固定化量を得やすい。本プロトコールでは、蛋白質のビオチン化に始まり、実際のスクリーニングを行うに至るまでの一連の実験手法・チェックポイントをご紹介する。
装置・機器・試薬
装置
- Biacore 8K(T200,T100 も可。しかしスループット性は下がる)(GE Healthcare)
- 遠心機(各社)
- スターラー(各社)
器具
- 透析膜(各社)
- 恒温インキュベータ(各社)
- 96 well deep well plate(Greiner,型番650201など)
- 384 well deep well plate(Greiner,型番781280など)
- プレートシール(GE Healthcare)
- Sensor Chip SA(GE Healthcare)
- AMICON(Millipore)
試薬
- BirA biotin-protein ligase standard reaction kit(Avidity 社)(Biomix-A、Biomix-B、BirA)
- 水酸化ナトリウム(各社)
- 塩化ナトリウム(各社)
- イソプロパノール(各社)
- Tween20(各社)
- 化合物ライブラリー(各社)
材料
- Avi-tag を付加した精製蛋白質
- 固定化蛋白質に対するポジティブコントロールとなる化合物
実験手順
- Avi-tag 付き蛋白質へのビオチン化反応
- 1-1. Avi-tag 付き蛋白質の準備
- 1-2. 反応バッファーへの透析
- 1-3. ビオチン化反応
- 1-4. SPR への固定化に向けた脱ビオチン用透析
- センサーチップへの固定化
- アッセイ系の評価(Z’ factor の算出)
- 化合物スクリーニングの実施
- 4-1. スクリーニング用メソッドの実施
- 4-2. 実験結果の解析
- 4-3. ヒットクライテリアの設定
- 結合レスポンスの値
- センサグラムの形状
- ヒット化合物の濃度依存性実験(2次スクリーニング)の実施
- 2次スクリーニング用メソッドの実施
- 実験結果の解析
実験の詳細
1)Avi-tag 付き蛋白質へのビオチン化反応
1-1)Avi-tag 付き蛋白質の準備
ビオチン化反応は Avi-tag 配列(GLNDIFEAQKIEWHE®)中で特異的に進むため、まずは標的蛋白質に Avi-tag を付加する必要がある。Avi-tag は15アミノ酸残基からなるタグであり、筆者は一般的な mutagenesis を用いて標的蛋白質に付加した。Mutagenesis のプロトコールは本プロトコールの趣旨とは外れるため割愛する。大腸菌発現系を用いて Avi-tag 付き蛋白質を発現し、Ni-NTA アフィニティカラムによる粗精製とサイズ排除クロマトグラフィー(SEC)による最終精製を経た蛋白質サンプルを以降のビオチン化反応に使用している。
1-2)反応バッファーへの透析
ビオチン化反応は 10 mM Tris-HCl,50 mM NaCl(pH 8.0)のバッファー条件下で行っている。塩濃度は低いほど反応に良いと言われている8。SEC 後のモノマー画分を反応に用いる濃度まで濃縮し、上記バッファーに4℃,16 h 透析する。
1-3)ビオチン化反応
上記反応バッファーに透析後の蛋白質を 40 μM ほどまで AMICON を用いて濃縮し、下記反応液組成に従ってビオチン化反応液を作成する。筆者はいつも1.5 mL エッペンチューブに反応液を作成している。
| Biomix-A | 100 μL |
| Biomix-B | 100 μL |
| 1.0 mg/mL BirA | 10 μL |
| 40 μM protein | 800 μL |
| To | 1010 μL |
反応液を作成する際、ビオチンリガーゼである BirA は直前まで氷上で取扱う。筆者はいつもBiomix-A,Biomix-B,protein,BirA の順番で混合している。作成した反応液は30℃,60 min incubation し、その後4℃で一晩保管してから次の手順に進めている。反応自体は30℃,60 min の条件で完了するとされている。
1-4)SPR への固定化に向けた脱ビオチン用透析
上記反応溶液を SPR 用バッファーへ透析する。これはビオチン化反応溶液中に残っている未反応のビオチンを除去するためのものである。この透析が不十分だと固定化量に影響が出る可能性がある。筆者はいつも 1010 μL の反応溶液を 1 L の SPR 用バッファーに対し4℃,16 h で透析したのちに SPR での固定化に用いている。この時、ビオチン化反応前の蛋白質溶液とビオチン化反応後の蛋白質溶液を 1:1 で混合したサンプルを 10 μL ほど作製し SDS-PAGE(15%アクリルアミド含有ゲル、CBB 染色)を行うと、ビオチン化された蛋白質はそうでない蛋白質より若干分子量の大きい位置にバンドが現れる9。ビオチン化反応が進行したかどうかはこれによって確認することができる(勿論、MALDI-TOFMS でビオチン化反応の進行を追うのも有効である)。
2)センサーチップへの固定化
SPR 測定は GE Healthcare 社の Biacore を使用する。各機器のコントロールソフトウェアには Sensor Chip SA への固定化メソッドが既に組まれており、そのメソッドの Instruction に従う。GE Healthcare 社の製品マニュアルでは pM オーダーの蛋白質濃度があればよいとされているが、筆者はいつも 10 μM ほどに調製したビオチン化反応後蛋白質溶液を固定化に用いている。固定化直前に4℃,20,000 G,10 min 遠心処理を行う。固定化メソッドでは最初に 50 mM NaOH,1 M NaCl によるセンサーチップのウォッシュが始まる。この時、アクティブフローセルのセンサグラムはセンサーチップ表面のコーティングがはがれることに由来して負に転ずる(図1)。その後ビオチン化蛋白質の固定化が行われ、最後に 50 mM NaOH,1 M NaCl,50%イソプロパノールによる蛋白質を固定化したフローセル以外のウォッシュを経てメソッドが完了する。Biacore 8K ではものの13分程度で固定化が完了する。
ここで、低分子リガンドのスクリーニングにはある程度の蛋白質の固定化量が要求されることに留意されたい。例えば、分子量 200 Da の低分子の結合を分子量 20,000 Da の蛋白質に対して 20 RU の結合レスポンスで検出したい場合、必要な固定化量は分子量の比より 2,000 RU と計算される(Biacore シリーズの検出限界は~1 RU レベルまで高性能化されているが、筆者は最低でもスクリーニング用途では 10 RU は検出できるように固定化量を稼ぐことで後のアッセイ系の評価がスムーズに進むと考える)。実際には、固定化した蛋白質の内何割かは失活しているため固定化後に得られた結合レスポンスに対し活性保持率を乗じた値を “真の固定化量” とせねばならない。さらに、固定化後一定時間バッファーを流し続け、固定化量が減少しないかを確認する必要がある。Biacore 8K を用いた低分子スクリーニングでは、前述したように384 well プレート1枚で6時間ほどの測定時間を要する。仮に、“真の固定化量” が 2000 RU 得られていたとしても、例えば 100 sec で 2 RU ずつ真の固定化量が減少してゆく場合、6時間後には 432 RU ほど固定化量が減少することになる。この減少量も考慮して必要な固定化量に達しているかを確認する必要がある。
3)アッセイ系の評価(Z’ factor の算出)
必要な固定化量が獲得できたら、今度は SPR のアッセイ系が低分子リガンドのスクリーニングの系として妥当かどうかを検証する。その際、スクリーニング系の妥当性を表す指標として用いられる Z’ factor を算出する10。Z’ factor の算出に当たって、結合が必ず得られるポジティブコントロールと結合が得られないネガティブコントロール(Running buffer)を用意し、図2a のようなプレートを作成する。化合物ライブラリー中の各化合物は基本的には DMSO に溶解されていることが多いので、Z’ factor の算出も DMSO 存在下で実施する。また、アッセイ中には定期的に Solvent correction(サンプルと running buffer 中のDMSO 濃度の差に由来するバルクレスポンスのフローセル間のずれを補正する手法)を実施する。筆者の場合は Running buffer に 5% DMSO を添加しており、Solvent correction は 4.5~6% DMSO で4点行っている。図2のプレートの各結合レスポンスを用いて Z’ factor を算出する(図2b)。この様にして得られたポジティブコントロールとネガティブコントロールの結合レスポンスより算出したZ’ factor が0.5以上であれば系として妥当だと言える。尚、このステップはポジティブコントロールがないと実施できない。
4)化合物スクリーニングの実施
4-1)スクリーニング用メソッドの実施
Z’ factor が無事0.5を上回れば、いよいよ低分子リガンドのスクリーニングを開始できる。化合物スクリーニングのメソッドを作成する場合でも、3)で述べた Solvent correction は勿論、加えて定期的にポジティブコントロールとネガティブコントロールの結合レスポンスを確認することが望まれる。これにより、固定化した蛋白質の活性を常に確認しながらスクリーニングを進めることができる。DMSO が吸湿性を持つため、化合物ライブラリーのプレート作成に際してはなるべく迅速な作業を心がける。例えば化合物終濃度 100 μM、5% DMSO で 70 μL 各化合物を調製する場合、予め384ウェルにはDMSO に溶解した 2 mM の各化合物を 3.5 μL 添加しておき、そこへ 66.5 μL の DMSO 不含 Running buffer を添加してゆく。添加の際には十分ピペッティングをし、吸湿による DMSO 濃度のブレを防ぐためピペッティングが終わったウェルから徐々にシールを貼って行く。全ウェルの調製が終わったらプレート遠心機で気泡を取り除き、スクリーニングに用いる。ライブラリー中に含まれている化合物には光による分解を受けるものが混ざっている可能性があるので、スクリーニングは遮光状態で行うのが安全である(Biacore のサンプルコンパートメントの窓部をアルミ等で覆う)。
4-2)実験結果の解析
得られた各化合物の結合レスポンスに対しては、ポジティブコントロールの結合レスポンスのサイクル依存的な変化を100%、ネガティブコントロールの結合レスポンスのサイクル依存的な変化を0%にノーマライズする “Adjustment for control” によって固定化した蛋白質の活性低下に伴う各化合物の結合レスポンスの差を均一化できる(図3a)。また、各化合物の分子量が分かっている場合は “Adjustment for molecular weight” によって各化合物の分子量による結合レスポンスの差も均一化できる。また筆者は化合物結合のレポートポイントとして “Binding_late” を使用している。Binding_late は化合物をセンサーチップ上にインジェクトし終わる直前での結合レスポンスを表す指標であり、一般的には箱型のセンサグラムとなるはずの低分子の蛋白質への結合を検出するには適したものだと筆者は考えている。
4-3)ヒットクライテリアの設定
ヒット化合物を選定する際にはヒットクライテリアを設定しなければならない。化合物スクリーニングにおいて絶対的な設定方法が決まっているわけではないが、ひとつの考え方を記す。
結合レスポンスの値
ヒットクライテリアの指標となる値の一つに結合レスポンスがある。例えば、先ほど述べた “真の固定化量” が 2,000 RU 得られており、分子量 20,000 Da の蛋白質に対し 200 Da ほどの低分子の結合を化合物の終濃度 100 μM で見る場合を想定する。もしこの化合物濃度が化合物と蛋白質の解離定数付近である場合、最大結合量 20 RU の内半分の 10 RU が検出されるはずである。換言すれば、仮に解離定数が 100 μM ほどの化合物を選択したければ 10 RU という値が一つのクライテリアとなる。逆に、この例において結合レスポンスが最大結合量よりも圧倒的に大きく(例えば 200 RU ほど)得られた場合は化合物の非特異的な結合、あるいは蛋白質中にいくつかの化合物結合部位があることが疑われる。こうした化合物はスクリーニング初期段階では拾い落としを防ぐという意味で続くアッセイに用いてもよいが、擬陽性である可能性が高いと言える。
センサグラムの形状
一般に、スクリーニング段階において低分子と蛋白質の相互作用(解離定数にして 1 mM~1 μM オーダー)を検出する際のセンサグラムの多くは箱型に近くなる(図3b)。これは経験上 kon が 103 M-1s-1 オーダー以上になるため上記の解離定数レンジでは必然的に koff が比較的速く解離する範囲の値になる結果といえそうである。逆に言えば、低分子にも拘わらず見かけ上の kon や koff が極めて遅い化合物は非特異的な結合を示す化合物と判断することがある(図3c)。先ほどと同様、スクリーニング初期段階ではヒット化合物にカウントしてもよいが、注意が必要である。
5)ヒット化合物の濃度依存性実験(2次スクリーニング)の実施
5-1)2次スクリーニング用メソッドの実施
1次スクリーニングで得られたヒット化合物については、続いて化合物濃度依存的な結合活性を示すかどうかを確認し擬陽性の排除を行う。1次スクリーニングの時と同様、Solvent correction は定期的にメソッドの中に組み込む。筆者はいつも各化合物に関して、濃度ゼロ以外に少なくとも6点の濃度勾配を設けて濃度依存性を確認している。濃度依存性確認の際にはゼロ以外のいずれかの濃度一点を最後のサイクルで繰り返し測定し、サイクル間で結合レスポンスに差がないことを確認する。これによりメソッド実施中の固定化蛋白質の失活がないかどうかを確認することができる。濃度勾配の設定は、解離定数の0.1~10倍程度の範囲で行うのが理想だと言われている。しかし、スクリーニングの場合そもそも解離定数が分かっていないことも多く、また化合物の溶解性によっては高い濃度設定が困難なことも予想される。筆者はいつも1 次スクリーニングの際の化合物濃度をまたぐようになるべく上記条件に近い濃度域で実施している。
5-2)実験結果の解析
得られた各化合物の結合レスポンスに関しては、Biacore の Evaluation software を使用して解離定数や速度論パラメータの算出を行う。Evaluation software では Kinetics 解析と Affinity 解析の2種類が存在する。Kinetics 解析では速度論パラメータも得られるが、センサグラムの形状が箱型の場合は Affinity 解析を行う(箱型のセンサグラムでは添加濃度における結合解離速度が速すぎるために速度論パラメータを定量的に議論できないため)。上述した通り、低分子の蛋白質に対する結合レスポンスは原則箱型となるため、実際には Affinity 解析を選択することになるはずである。Affinity 解析においては化合物濃度に対する結合レスポンスから Scatchard plot を作成し、解離定数と最大結合量を示す RMAX を得る。この時得られた RMAX 値が再び蛋白質と低分子の分子量及び、“真の固定化量” から計算される RMAX の理論値と大差ない(例えばオーダーが同じ)値であるか確認する。この条件を満たしている場合、その化合物は2次スクリーニングのヒット化合物といえる(図4a)。逆に、仮に理論値に対して実測値があまりに大きい場合は非特異的な結合を示す擬陽性の化合物だと考えることができる(図4b)。また、得られた解離定数は一番高い化合物濃度の半分以下の濃度であることが望ましい。仮にこの条件を満たさない場合、その解離定数の信頼性は低く、化合物の濃度範囲を変更して再測定する必要がある。ただし、化合物の溶解性から高濃度添加が困難な場合は低濃度帯の plot を用いてポジティブコントロールのレスポンスと該当する化合物の分子量から算出される RMAX の値を定数(constant Rmax)とした平衡値解析モデルで解析することも可能である。
工夫とコツ
Avi-tag の付加に関して
Avi-tag を N 末端に付加するか、C 末端に付加するかは標的蛋白質の性質に応じて判断されるべきで、原則は存在しない。また、筆者らの経験上、標的蛋白質と Avi-tag の間にリンカー(筆者は G3S リンカーを使用している)を挟むかどうかでビオチン化反応効率が顕著に変わることもあった。ビオチン化反応が安定して進むコンストラクトの検討が場合によっては必要である。
Avi-tag 付き蛋白質の取り扱い
筆者の経験上は Avi-tag を付けた蛋白質の発現パターンや精製条件はタグなしの場合と変わらず行えた。
DMSO 存在下での蛋白質活性
化合物をバッファー中に溶かすためにはDMSO が用いられる。DMSO 含有バッファーにおいてはある割合の蛋白質が失活する可能性がある。筆者は DMSO 存在下でのポジティブコントロールの解離定数の算出や Z’ factor の算出を行うことで自身の蛋白質が DMSO に耐えうるかどうかを予め確認している。
DMSO のバルク効果を抑える工夫(保管、攪拌)
DMSO を含むバッファー・プレートの調製は先に述べた通り十分注意する。細心の注意を払う際、筆者は以下の点に留意している。
- あるメソッドで使用するプレートと Running buffer の中にロットの違う DMSO が混ざることを避ける。
- DMSO は遮光し、デシケータで保管する。
- ウェル内では溶液が攪拌されにくい。筆者はウェルに入れる溶液の半分以上の容量でのピペッティングを励行している。
活性保持率の算出方法
固定化した蛋白質の活性保持率は、理論的な RMAX に対する実測の RMAX の比で求められる。実測の RMAX はリガンドの濃度依存的な結合レスポンスから Kinetics 解析や Affinity 解析によって算出される。
実験の安全
- 水酸化ナトリウムは強塩基なので、取り扱いには十分注意する。
- イソプロパノールは引火性の有機溶媒なので、取り扱いには十分注意する。
- Biacore は流路のつまりが故障の大きな原因のひとつである。化合物や蛋白質の凝集体は流路を詰まらせ、パーツの交換等につながる可能性がある。蛋白質は実験前に必ず遠心処理をし、化合物の高濃度での使用は控える。
文献
- Frostell, A., et al., Methods Mol. Biol., 1008, 139–165 (2013)
- Folmer R. H., Drug Discov. Today, 21, 491–498 (2016)
- Tashiro S., et al., ACS Chem. Biol., 13, 2783–2793 (2018)
- Bai Y., et al., Protein Cell, 6, 924–928 (2015)
- Senoo A., et al., Chem. Commun., 54, 5350–5353 (2018)
- Kobe A., et al., J. Med. Chem., 56, 2155–2159 (2013)
- Giannetti A. M., Methods Enzymol., 493, 169–218 (2011)
- Cull M. G., et al., Methods Enzymol., 326, 430–440 (2000)
- Yifeng L., et al., Protein Expres. Purif., 82, 162–165 (2012)
- 長野哲雄 編、創薬化学 –メディシナルケミストへの道–, 東京化学同人, 83–84 (2018)
謝辞
本プロトコールを作成するにあたり、GE ヘルスケア・ジャパン株式会社の三谷知也様にサポートを賜りました。また、プロトコール作成に関連する研究は JSPS 特別研究員奨励費 JP19J14451 の助成を受けたものです。心より感謝申し上げます。
変更履歴
| 変更日 | 変更内容 | 変更前の PDF |
|---|---|---|
| 2020/05/12 | 「実験の詳細」に記載された Avi-tag の配列の11番目の「L」を「I」に変更、文献7を変更、新たな文献8を追加、文献8, 9の文献番号をそれぞれ9, 10に変更 | 変更前の PDF |
-
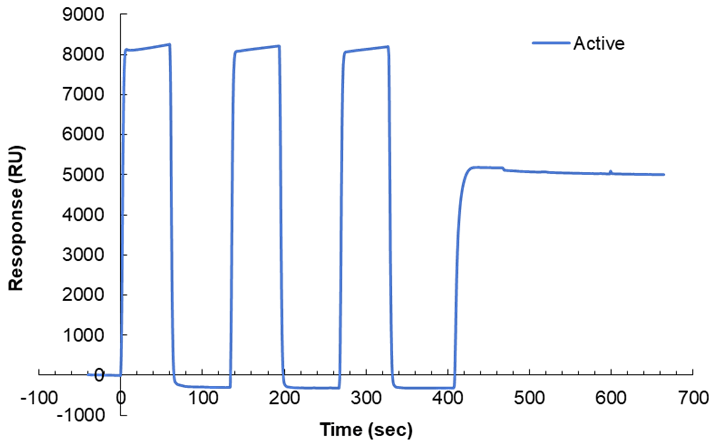
図1:Sensor Chip SA への固定化プロファイル。Active セルのセンサグラムは、3回のウォッシュ後に蛋白質の固定化を行う。 -
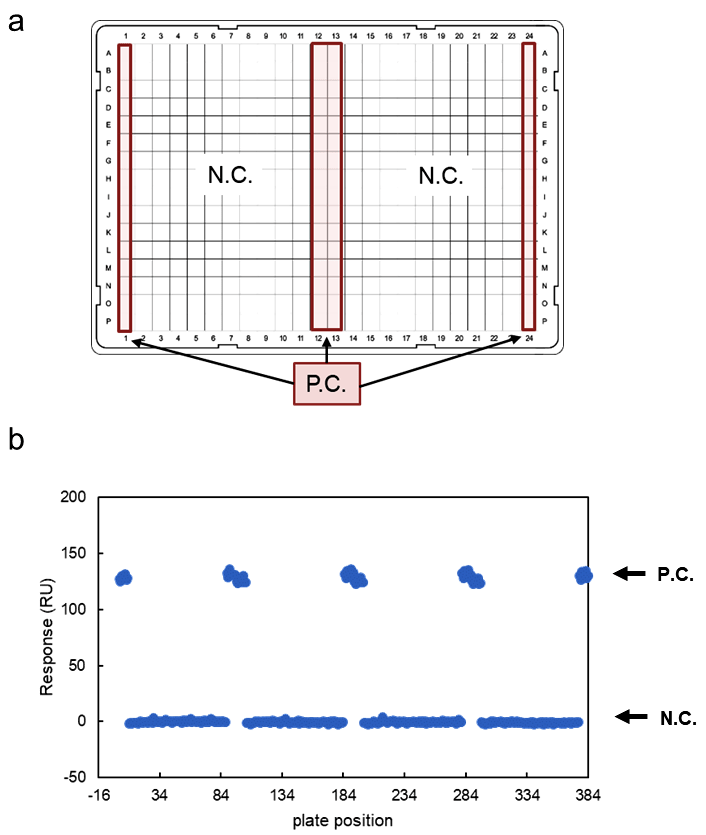
図2:Z’ factor の算出。(a)プレート作製例(b)このプレートに対して行ったポジティブコントロール(P.C.)のリガンドとネガティブコントロール(N.C.)の結合レスポンス。この生データから算出した Z’ factor は0.89と良好な値を示した。 -
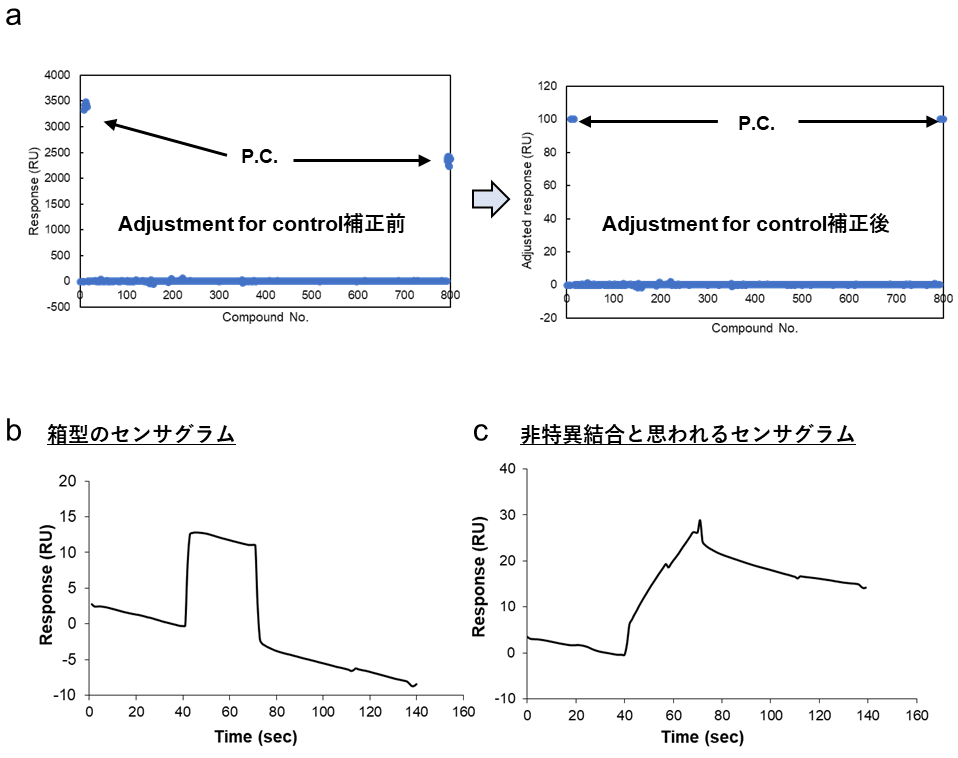
図3:低分子スクリーニングの生データ事例。(a)Adjustment for control によるレスポンスの均一化事例。補正後は P.C. の結合量を一律 100 RU として、各サンプルの結合レスポンスを取得することになる。(b)いわゆる箱型と呼ばれるセンサグラム。一般的な蛋白質-低分子間相互作用はこのような形になることが多い。(c)kon,koff ともに極めて遅く、非特異的結合と判断されたセンサグラム。 -
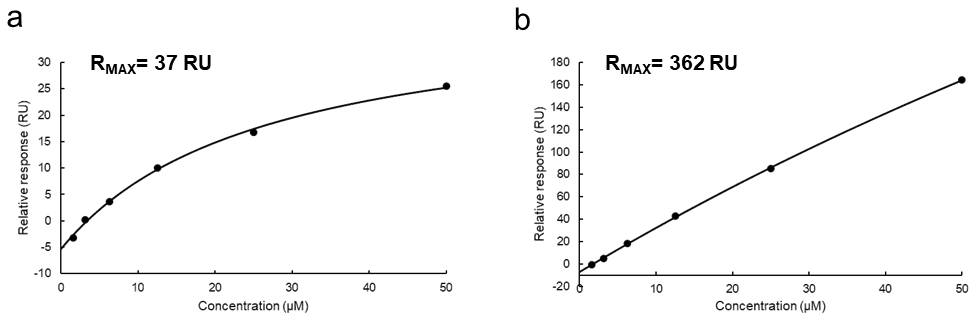
図4:結合レスポンスの濃度依存性試験で得られた Scatchard plot の事例。RMAX の理論値はいずれも 37 RU ほどの化合物の結合を確認している。(a)RMAX が理論値と大差なかったサンプルの Scatchard plot。高濃度の化合物領域では結合がほぼ飽和に達しており、2次スクリーニングではヒット化合物とみなせる。(b)RMAX が理論値よりも明らかに大きかったサンプルの Scatchard plot。本来曲線的に飽和に達するはずの結合値が直線的に増加して行っており、顕著に非特異的な結合を示す偽陽性の化合物であると考えられる。
概要
蛋白質に結合する低分子化合物(以下、低分子リガンド)は、創薬標的蛋白質に対する分子標的薬や機能未知の蛋白質に対するケミカルノックダウンの手法としての応用が期待される。蛋白質と低分子リガンドの相互作用を検出する手法には本プロトコールで取り上げる表面プラズモン共鳴(SPR)1 の他に、等温滴定型熱量測定(ITC)、示差走査蛍光測定(DSF,もしくはいわゆる Thermal Shift Assay)、核磁気共鳴分光法(NMR)、マイクロスケール熱泳動(MST)、バイオレイヤー干渉法(BLI)など様々な物理化学的手法の選択が可能である2。しかしながら、低分子リガンドを大量の分子種からなる化合物ライブラリーの中からハイスループットに選抜しようとした際、多くの手法は使用する蛋白質の量・検出感度・スループット性の問題から必ずしも最適とは言えない。それに対し、SPR は比較的スループット性を担保した(本プロトコールで紹介する手法では、384ウェルプレート1枚あたりおよそ6時間)装置を用いることにより、酵素・蛋白質–蛋白質間相互作用に関わる蛋白質・膜蛋白質といった様々な標的に対する低分子リガンドを選抜することができる3–6。SPR の相互作用検出原理は表面プラズモン共鳴を活用した質量変化であるため、蛋白質に対する低分子リガンドの結合を、エンタルピー駆動・エントロピー駆動等の相互作用様式によらず選抜することも可能である。また、SPR のセンサグラムからは蛋白質-リガンド相互作用における速度論パラメータを議論することも可能であるため、これらの情報を活用することで一連の研究プロセスの早い段階にて擬陽性の化合物を排除することも可能である。したがって SPR による低分子リガンド選抜は、有効な低分子スクリーニング技術の1つとして挙げられる。
イントロダクション
SPR による相互作用測定ではセンサーチップ上に標的となる蛋白質を固定化し、そこへ化合物を一定の流速で流してゆく。蛋白質の固定化には様々な手法が用いられているが、本プロトコールでは Sensor Chip SA に対してビオチン化した蛋白質の固定化法について記す。この手法が選択された背景には、低分子と蛋白質の相互作用を測定するのに「安定的なベースライン」と「十分な蛋白質固定化量」が欠かせないことが挙げられる。前者に関しては、例えば Sensor Chip Ni-NTA に対する His キャプチャーを用いた固定化法では、実験中にセンサーチップ上から蛋白質が乖離し正確な結合測定が行えない問題がしばしば散見される。後者に関しては、例えば Sensor Chip CM5, CM7 に対するアミンカップリング法では、固定化に際し蛋白質を酸性 pH にさらすことや、蛋白質が多点で固定化されることに由来する蛋白質の失活が重大な固定化量の低下を招くことがある。こうした問題を同時に解消する可能性があるのが、本プロトコールで採用した SA-biotin キャプチャー法である。Streptavidin とビオチンの高い親和性から、実験中の蛋白質のセンサーチップ上からの解離はほとんど起こらない。さらに、ビオチン化修飾は蛋白質の N 末端か C 末端を選択して Avi-tag を付加し、タグに対しビオチン1分子が特異的に修飾される酵素反応を使用する7–9ため多点での蛋白質の固定化も防ぐことができる。そのため蛋白質の活性を保った状態で十分な固定化量を得やすい。本プロトコールでは、蛋白質のビオチン化に始まり、実際のスクリーニングを行うに至るまでの一連の実験手法・チェックポイントをご紹介する。
装置・機器・試薬
装置
- Biacore 8K(T200,T100 も可。しかしスループット性は下がる)(GE Healthcare)
- 遠心機(各社)
- スターラー(各社)
器具
- 透析膜(各社)
- 恒温インキュベータ(各社)
- 96 well deep well plate(Greiner,型番650201など)
- 384 well deep well plate(Greiner,型番781280など)
- プレートシール(GE Healthcare)
- Sensor Chip SA(GE Healthcare)
- AMICON(Millipore)
試薬
- BirA biotin-protein ligase standard reaction kit(Avidity 社)(Biomix-A、Biomix-B、BirA)
- 水酸化ナトリウム(各社)
- 塩化ナトリウム(各社)
- イソプロパノール(各社)
- Tween20(各社)
- 化合物ライブラリー(各社)
材料
- Avi-tag を付加した精製蛋白質
- 固定化蛋白質に対するポジティブコントロールとなる化合物
実験手順
- Avi-tag 付き蛋白質へのビオチン化反応
- 1-1. Avi-tag 付き蛋白質の準備
- 1-2. 反応バッファーへの透析
- 1-3. ビオチン化反応
- 1-4. SPR への固定化に向けた脱ビオチン用透析
- センサーチップへの固定化
- アッセイ系の評価(Z’ factor の算出)
- 化合物スクリーニングの実施
- 4-1. スクリーニング用メソッドの実施
- 4-2. 実験結果の解析
- 4-3. ヒットクライテリアの設定
- 結合レスポンスの値
- センサグラムの形状
- ヒット化合物の濃度依存性実験(2次スクリーニング)の実施
- 2次スクリーニング用メソッドの実施
- 実験結果の解析
実験の詳細
1)Avi-tag 付き蛋白質へのビオチン化反応
1-1)Avi-tag 付き蛋白質の準備
ビオチン化反応は Avi-tag 配列(GLNDIFEAQKIEWHE®)中で特異的に進むため、まずは標的蛋白質に Avi-tag を付加する必要がある。Avi-tag は15アミノ酸残基からなるタグであり、筆者は一般的な mutagenesis を用いて標的蛋白質に付加した。Mutagenesis のプロトコールは本プロトコールの趣旨とは外れるため割愛する。大腸菌発現系を用いて Avi-tag 付き蛋白質を発現し、Ni-NTA アフィニティカラムによる粗精製とサイズ排除クロマトグラフィー(SEC)による最終精製を経た蛋白質サンプルを以降のビオチン化反応に使用している。
1-2)反応バッファーへの透析
ビオチン化反応は 10 mM Tris-HCl,50 mM NaCl(pH 8.0)のバッファー条件下で行っている。塩濃度は低いほど反応に良いと言われている8。SEC 後のモノマー画分を反応に用いる濃度まで濃縮し、上記バッファーに4℃,16 h 透析する。
1-3)ビオチン化反応
上記反応バッファーに透析後の蛋白質を 40 μM ほどまで AMICON を用いて濃縮し、下記反応液組成に従ってビオチン化反応液を作成する。筆者はいつも1.5 mL エッペンチューブに反応液を作成している。
| Biomix-A | 100 μL |
| Biomix-B | 100 μL |
| 1.0 mg/mL BirA | 10 μL |
| 40 μM protein | 800 μL |
| To | 1010 μL |
反応液を作成する際、ビオチンリガーゼである BirA は直前まで氷上で取扱う。筆者はいつもBiomix-A,Biomix-B,protein,BirA の順番で混合している。作成した反応液は30℃,60 min incubation し、その後4℃で一晩保管してから次の手順に進めている。反応自体は30℃,60 min の条件で完了するとされている。
1-4)SPR への固定化に向けた脱ビオチン用透析
上記反応溶液を SPR 用バッファーへ透析する。これはビオチン化反応溶液中に残っている未反応のビオチンを除去するためのものである。この透析が不十分だと固定化量に影響が出る可能性がある。筆者はいつも 1010 μL の反応溶液を 1 L の SPR 用バッファーに対し4℃,16 h で透析したのちに SPR での固定化に用いている。この時、ビオチン化反応前の蛋白質溶液とビオチン化反応後の蛋白質溶液を 1:1 で混合したサンプルを 10 μL ほど作製し SDS-PAGE(15%アクリルアミド含有ゲル、CBB 染色)を行うと、ビオチン化された蛋白質はそうでない蛋白質より若干分子量の大きい位置にバンドが現れる9。ビオチン化反応が進行したかどうかはこれによって確認することができる(勿論、MALDI-TOFMS でビオチン化反応の進行を追うのも有効である)。
2)センサーチップへの固定化
SPR 測定は GE Healthcare 社の Biacore を使用する。各機器のコントロールソフトウェアには Sensor Chip SA への固定化メソッドが既に組まれており、そのメソッドの Instruction に従う。GE Healthcare 社の製品マニュアルでは pM オーダーの蛋白質濃度があればよいとされているが、筆者はいつも 10 μM ほどに調製したビオチン化反応後蛋白質溶液を固定化に用いている。固定化直前に4℃,20,000 G,10 min 遠心処理を行う。固定化メソッドでは最初に 50 mM NaOH,1 M NaCl によるセンサーチップのウォッシュが始まる。この時、アクティブフローセルのセンサグラムはセンサーチップ表面のコーティングがはがれることに由来して負に転ずる(図1)。その後ビオチン化蛋白質の固定化が行われ、最後に 50 mM NaOH,1 M NaCl,50%イソプロパノールによる蛋白質を固定化したフローセル以外のウォッシュを経てメソッドが完了する。Biacore 8K ではものの13分程度で固定化が完了する。
ここで、低分子リガンドのスクリーニングにはある程度の蛋白質の固定化量が要求されることに留意されたい。例えば、分子量 200 Da の低分子の結合を分子量 20,000 Da の蛋白質に対して 20 RU の結合レスポンスで検出したい場合、必要な固定化量は分子量の比より 2,000 RU と計算される(Biacore シリーズの検出限界は~1 RU レベルまで高性能化されているが、筆者は最低でもスクリーニング用途では 10 RU は検出できるように固定化量を稼ぐことで後のアッセイ系の評価がスムーズに進むと考える)。実際には、固定化した蛋白質の内何割かは失活しているため固定化後に得られた結合レスポンスに対し活性保持率を乗じた値を “真の固定化量” とせねばならない。さらに、固定化後一定時間バッファーを流し続け、固定化量が減少しないかを確認する必要がある。Biacore 8K を用いた低分子スクリーニングでは、前述したように384 well プレート1枚で6時間ほどの測定時間を要する。仮に、“真の固定化量” が 2000 RU 得られていたとしても、例えば 100 sec で 2 RU ずつ真の固定化量が減少してゆく場合、6時間後には 432 RU ほど固定化量が減少することになる。この減少量も考慮して必要な固定化量に達しているかを確認する必要がある。
3)アッセイ系の評価(Z’ factor の算出)
必要な固定化量が獲得できたら、今度は SPR のアッセイ系が低分子リガンドのスクリーニングの系として妥当かどうかを検証する。その際、スクリーニング系の妥当性を表す指標として用いられる Z’ factor を算出する10。Z’ factor の算出に当たって、結合が必ず得られるポジティブコントロールと結合が得られないネガティブコントロール(Running buffer)を用意し、図2a のようなプレートを作成する。化合物ライブラリー中の各化合物は基本的には DMSO に溶解されていることが多いので、Z’ factor の算出も DMSO 存在下で実施する。また、アッセイ中には定期的に Solvent correction(サンプルと running buffer 中のDMSO 濃度の差に由来するバルクレスポンスのフローセル間のずれを補正する手法)を実施する。筆者の場合は Running buffer に 5% DMSO を添加しており、Solvent correction は 4.5~6% DMSO で4点行っている。図2のプレートの各結合レスポンスを用いて Z’ factor を算出する(図2b)。この様にして得られたポジティブコントロールとネガティブコントロールの結合レスポンスより算出したZ’ factor が0.5以上であれば系として妥当だと言える。尚、このステップはポジティブコントロールがないと実施できない。
4)化合物スクリーニングの実施
4-1)スクリーニング用メソッドの実施
Z’ factor が無事0.5を上回れば、いよいよ低分子リガンドのスクリーニングを開始できる。化合物スクリーニングのメソッドを作成する場合でも、3)で述べた Solvent correction は勿論、加えて定期的にポジティブコントロールとネガティブコントロールの結合レスポンスを確認することが望まれる。これにより、固定化した蛋白質の活性を常に確認しながらスクリーニングを進めることができる。DMSO が吸湿性を持つため、化合物ライブラリーのプレート作成に際してはなるべく迅速な作業を心がける。例えば化合物終濃度 100 μM、5% DMSO で 70 μL 各化合物を調製する場合、予め384ウェルにはDMSO に溶解した 2 mM の各化合物を 3.5 μL 添加しておき、そこへ 66.5 μL の DMSO 不含 Running buffer を添加してゆく。添加の際には十分ピペッティングをし、吸湿による DMSO 濃度のブレを防ぐためピペッティングが終わったウェルから徐々にシールを貼って行く。全ウェルの調製が終わったらプレート遠心機で気泡を取り除き、スクリーニングに用いる。ライブラリー中に含まれている化合物には光による分解を受けるものが混ざっている可能性があるので、スクリーニングは遮光状態で行うのが安全である(Biacore のサンプルコンパートメントの窓部をアルミ等で覆う)。
4-2)実験結果の解析
得られた各化合物の結合レスポンスに対しては、ポジティブコントロールの結合レスポンスのサイクル依存的な変化を100%、ネガティブコントロールの結合レスポンスのサイクル依存的な変化を0%にノーマライズする “Adjustment for control” によって固定化した蛋白質の活性低下に伴う各化合物の結合レスポンスの差を均一化できる(図3a)。また、各化合物の分子量が分かっている場合は “Adjustment for molecular weight” によって各化合物の分子量による結合レスポンスの差も均一化できる。また筆者は化合物結合のレポートポイントとして “Binding_late” を使用している。Binding_late は化合物をセンサーチップ上にインジェクトし終わる直前での結合レスポンスを表す指標であり、一般的には箱型のセンサグラムとなるはずの低分子の蛋白質への結合を検出するには適したものだと筆者は考えている。
4-3)ヒットクライテリアの設定
ヒット化合物を選定する際にはヒットクライテリアを設定しなければならない。化合物スクリーニングにおいて絶対的な設定方法が決まっているわけではないが、ひとつの考え方を記す。
結合レスポンスの値
ヒットクライテリアの指標となる値の一つに結合レスポンスがある。例えば、先ほど述べた “真の固定化量” が 2,000 RU 得られており、分子量 20,000 Da の蛋白質に対し 200 Da ほどの低分子の結合を化合物の終濃度 100 μM で見る場合を想定する。もしこの化合物濃度が化合物と蛋白質の解離定数付近である場合、最大結合量 20 RU の内半分の 10 RU が検出されるはずである。換言すれば、仮に解離定数が 100 μM ほどの化合物を選択したければ 10 RU という値が一つのクライテリアとなる。逆に、この例において結合レスポンスが最大結合量よりも圧倒的に大きく(例えば 200 RU ほど)得られた場合は化合物の非特異的な結合、あるいは蛋白質中にいくつかの化合物結合部位があることが疑われる。こうした化合物はスクリーニング初期段階では拾い落としを防ぐという意味で続くアッセイに用いてもよいが、擬陽性である可能性が高いと言える。
センサグラムの形状
一般に、スクリーニング段階において低分子と蛋白質の相互作用(解離定数にして 1 mM~1 μM オーダー)を検出する際のセンサグラムの多くは箱型に近くなる(図3b)。これは経験上 kon が 103 M-1s-1 オーダー以上になるため上記の解離定数レンジでは必然的に koff が比較的速く解離する範囲の値になる結果といえそうである。逆に言えば、低分子にも拘わらず見かけ上の kon や koff が極めて遅い化合物は非特異的な結合を示す化合物と判断することがある(図3c)。先ほどと同様、スクリーニング初期段階ではヒット化合物にカウントしてもよいが、注意が必要である。
5)ヒット化合物の濃度依存性実験(2次スクリーニング)の実施
5-1)2次スクリーニング用メソッドの実施
1次スクリーニングで得られたヒット化合物については、続いて化合物濃度依存的な結合活性を示すかどうかを確認し擬陽性の排除を行う。1次スクリーニングの時と同様、Solvent correction は定期的にメソッドの中に組み込む。筆者はいつも各化合物に関して、濃度ゼロ以外に少なくとも6点の濃度勾配を設けて濃度依存性を確認している。濃度依存性確認の際にはゼロ以外のいずれかの濃度一点を最後のサイクルで繰り返し測定し、サイクル間で結合レスポンスに差がないことを確認する。これによりメソッド実施中の固定化蛋白質の失活がないかどうかを確認することができる。濃度勾配の設定は、解離定数の0.1~10倍程度の範囲で行うのが理想だと言われている。しかし、スクリーニングの場合そもそも解離定数が分かっていないことも多く、また化合物の溶解性によっては高い濃度設定が困難なことも予想される。筆者はいつも1 次スクリーニングの際の化合物濃度をまたぐようになるべく上記条件に近い濃度域で実施している。
5-2)実験結果の解析
得られた各化合物の結合レスポンスに関しては、Biacore の Evaluation software を使用して解離定数や速度論パラメータの算出を行う。Evaluation software では Kinetics 解析と Affinity 解析の2種類が存在する。Kinetics 解析では速度論パラメータも得られるが、センサグラムの形状が箱型の場合は Affinity 解析を行う(箱型のセンサグラムでは添加濃度における結合解離速度が速すぎるために速度論パラメータを定量的に議論できないため)。上述した通り、低分子の蛋白質に対する結合レスポンスは原則箱型となるため、実際には Affinity 解析を選択することになるはずである。Affinity 解析においては化合物濃度に対する結合レスポンスから Scatchard plot を作成し、解離定数と最大結合量を示す RMAX を得る。この時得られた RMAX 値が再び蛋白質と低分子の分子量及び、“真の固定化量” から計算される RMAX の理論値と大差ない(例えばオーダーが同じ)値であるか確認する。この条件を満たしている場合、その化合物は2次スクリーニングのヒット化合物といえる(図4a)。逆に、仮に理論値に対して実測値があまりに大きい場合は非特異的な結合を示す擬陽性の化合物だと考えることができる(図4b)。また、得られた解離定数は一番高い化合物濃度の半分以下の濃度であることが望ましい。仮にこの条件を満たさない場合、その解離定数の信頼性は低く、化合物の濃度範囲を変更して再測定する必要がある。ただし、化合物の溶解性から高濃度添加が困難な場合は低濃度帯の plot を用いてポジティブコントロールのレスポンスと該当する化合物の分子量から算出される RMAX の値を定数(constant Rmax)とした平衡値解析モデルで解析することも可能である。
工夫とコツ
Avi-tag の付加に関して
Avi-tag を N 末端に付加するか、C 末端に付加するかは標的蛋白質の性質に応じて判断されるべきで、原則は存在しない。また、筆者らの経験上、標的蛋白質と Avi-tag の間にリンカー(筆者は G3S リンカーを使用している)を挟むかどうかでビオチン化反応効率が顕著に変わることもあった。ビオチン化反応が安定して進むコンストラクトの検討が場合によっては必要である。
Avi-tag 付き蛋白質の取り扱い
筆者の経験上は Avi-tag を付けた蛋白質の発現パターンや精製条件はタグなしの場合と変わらず行えた。
DMSO 存在下での蛋白質活性
化合物をバッファー中に溶かすためにはDMSO が用いられる。DMSO 含有バッファーにおいてはある割合の蛋白質が失活する可能性がある。筆者は DMSO 存在下でのポジティブコントロールの解離定数の算出や Z’ factor の算出を行うことで自身の蛋白質が DMSO に耐えうるかどうかを予め確認している。
DMSO のバルク効果を抑える工夫(保管、攪拌)
DMSO を含むバッファー・プレートの調製は先に述べた通り十分注意する。細心の注意を払う際、筆者は以下の点に留意している。
- あるメソッドで使用するプレートと Running buffer の中にロットの違う DMSO が混ざることを避ける。
- DMSO は遮光し、デシケータで保管する。
- ウェル内では溶液が攪拌されにくい。筆者はウェルに入れる溶液の半分以上の容量でのピペッティングを励行している。
活性保持率の算出方法
固定化した蛋白質の活性保持率は、理論的な RMAX に対する実測の RMAX の比で求められる。実測の RMAX はリガンドの濃度依存的な結合レスポンスから Kinetics 解析や Affinity 解析によって算出される。
実験の安全
- 水酸化ナトリウムは強塩基なので、取り扱いには十分注意する。
- イソプロパノールは引火性の有機溶媒なので、取り扱いには十分注意する。
- Biacore は流路のつまりが故障の大きな原因のひとつである。化合物や蛋白質の凝集体は流路を詰まらせ、パーツの交換等につながる可能性がある。蛋白質は実験前に必ず遠心処理をし、化合物の高濃度での使用は控える。
文献
- Frostell, A., et al., Methods Mol. Biol., 1008, 139–165 (2013)
- Folmer R. H., Drug Discov. Today, 21, 491–498 (2016)
- Tashiro S., et al., ACS Chem. Biol., 13, 2783–2793 (2018)
- Bai Y., et al., Protein Cell, 6, 924–928 (2015)
- Senoo A., et al., Chem. Commun., 54, 5350–5353 (2018)
- Kobe A., et al., J. Med. Chem., 56, 2155–2159 (2013)
- Giannetti A. M., Methods Enzymol., 493, 169–218 (2011)
- Cull M. G., et al., Methods Enzymol., 326, 430–440 (2000)
- Yifeng L., et al., Protein Expres. Purif., 82, 162–165 (2012)
- 長野哲雄 編、創薬化学 –メディシナルケミストへの道–, 東京化学同人, 83–84 (2018)
謝辞
本プロトコールを作成するにあたり、GE ヘルスケア・ジャパン株式会社の三谷知也様にサポートを賜りました。また、プロトコール作成に関連する研究は JSPS 特別研究員奨励費 JP19J14451 の助成を受けたものです。心より感謝申し上げます。
変更履歴
| 変更日 | 変更内容 | 変更前の PDF |
|---|---|---|
| 2020/05/12 | 「実験の詳細」に記載された Avi-tag の配列の11番目の「L」を「I」に変更、文献7を変更、新たな文献8を追加、文献8, 9の文献番号をそれぞれ9, 10に変更 | 変更前の PDF |
