

概要
円二色性(CD)は、ペプチド鎖のコンフォメーションの違いを敏感に反映するため、蛋白質の二次構造の含量解析に利用されている(1,2)。またCDは、多様な実験条件下で測定できるため、X線結晶学やNMR法では難しい膜蛋白質や非天然状態の蛋白質の構造解析に有力である。しかし、通常のCD分光法は、短波長側の測定範囲が遠紫外領域である190 nm程度にとどまり、得られる二次構造情報に限界がある。真空紫外円二色性(VUVCD)分光法は、放射光を用いることで、真空紫外(VUV)領域の高エネルギー遷移に基づいたCDを検出することができ、またバイオインフォマティクス技術を取り入れたCD解析から、二次構造の含量や本数の算出に加え、アミノ酸残基レベルでの二次構造の位置予測が可能である。本稿では、広島大学放射光科学研究センター(HiSOR) のVUVCD実験のプロトコールとして、VUV領域で正確なCDスペクトルを得るための蛋白質溶液の調製及び測定方法、そして様々な二次構造情報を得るためのスペクトルの解析方法を紹介する。
イントロダクション
VUVCD測定が可能な放射光CD装置は、欧米アジアの放射光施設で10機以上開発されている(3)。我が国では、HiSOR(図1)においてCDスペクトルが140 nmまで測定できるVUVCD分散計(図2)が開発されており、現在、全国共同利用装置として稼働している。VUVCD実験を行うための手順は、基本的にはCD実験と同様であるが(4)、放射光を利用するための申請書を提出する点、またサンプルの調製や測定方法そして二次構造の解析方法で異なる点がある。以下に、実験手順を4つのSTEPに分けて説明する。
装置・器具・試薬
- 真空紫外円二色性分散計(HiSOR BL-15)
- 組み立て式光学セル(HiSOR BL-15)
- 分光光度計(例えば、JASCO V-630BIO等)
- 遠心分離機
- メンブレンフィルター(孔径: 0.2~20μm)
- 解析ソフト(OriginやMicrosoft Excel等)
- 蛋白質二次構造解析プログラム(CDPro: http://lamar.colostate.edu/~sreeram/SELCON3/ 及びDichroWeb: http://dichroweb.cryst.bbk.ac.uk/html/home.shtml を参照)
実験手順
STEP1 研究課題申請書の提出
・研究課題の申請
STEP2 サンプルの準備
・蛋白質溶液の調製
・蛋白質濃度の決定
STEP3 スペクトル測定の実施
・測定の準備
・測定の開始
STEP4 スペクトル解析(二次構造解析)
・モル楕円率の計算
・スペクトル解析
実験の詳細
STEP1 研究課題申請書の提出
・研究課題の申請
HiSORでVUVCD測定を行うには、研究内容や実験期間等を記した研究課題申請書を提出する必要がある。申請書の提出時期は、毎年1月末頃である。申請方法あるいは実験の技術的な詳細は、HiSORの担当者と相談することができる。申請書の作成から実験に至るまでの手続の流れは、HiSORのホームページ(http://www.hsrc.hiroshima-u.ac.jp/format.htm)で紹介されている。
STEP2 サンプルの準備
・蛋白質溶液の調製
スペクトルの精度は、二次構造解析の結果に影響するため、測定に適した溶液調製が必要である。蛋白質の純度は、質量分析法やSDSポリアクリルアミドゲル電気泳動等を用いて、95%以上であることを確かめる。VUVCD測定では、VUV領域における水の吸収を抑えるため、光路長が極短 (1.3~100μm)な光学セルを使用する。そのため、測定に要する蛋白質溶液は微量(1~30μl)であるが、通常のCD測定に比べ高い濃度が必要である。濃度の目安は、使用する光路長や蛋白質の種類に依存するが、50~250μM(重量パーセント濃度では、0.05~0.5%)程度が目安である。また、会合体による光散乱は、スペクトルのベースラインシフトの原因となるため、遠心分離機(例えば、20,400G・20分間)やろ過(例えば、孔径0.2~20μmのメンブレンフィルター)等で、測定前に蛋白質溶液から取り除く。
VUV領域に大きな吸収を持つ塩あるいは緩衝液は、スペクトルのS/N比の低下や測定領域の拡張の障害となるため、使用を避けるか濃度を低く抑える。塩化物イオンを含むNaCl(あるいはCaCl2)は、蛋白質の安定性・溶解性・不活性化等の理由で必須な場合を除いて使用を避けるか、イオン濃度が5 mM以下になるように抑える。塩化物イオンを含まないNaFは、150 mM程度でも吸収が少ないため、NaClの代わりに利用できる。pH 8.0~9.0の範囲の溶液調製では、NaOHの使用は避けるべきで、Tris緩衝液(< 50 mM)の利用が好ましい。Tris緩衝液を作成する時は、HClの代わりに、リン酸や硫酸を用いた方が良い。pH5.5~8.0の範囲では、リン酸緩衝液(< 50 mM)が最も適しており、Tris緩衝液や吸収の大きいHEPES緩衝液の使用は控える。また、pH5.5~3.5では、酢酸緩衝液(< 50 mM)が最も良い。pH3.5~2.0では、HClの利用が好ましく、吸収の大きいクエン酸緩衝液の使用は控える。その他の溶媒として、DTTは数 mMでは測定にほとんど影響が無いが、EDTAは0.5 mMでもスペクトルのS/N比に影響するので、できれば使用を避けたい。有機溶媒であるアセトニトリル、メタノール、エタノール、ブタノール、トリフルオロエタノール、ヘキサフルオロイソプロパノールは、VUV領域の測定に適しているが、メタノールを使用した場合、180~185 nmまでのCD測定が限界である。
以上を参考にして、塩や緩衝液の種類や濃度を決定する。可能であれば、蛋白質溶液を調製する前に、使用する溶媒のVUV領域での吸収を測定することをお勧めする。
・蛋白質濃度の決定
アミノ酸配列が既知な蛋白質の濃度を決定する最良の方法は、アミノ酸分析である。この分析では、一定量の溶液中にある蛋白質を、6 N HCl・110℃の下、24時間保持し、完全にアミノ酸単体に加水分解させる。続いて、アミノ酸分析装置により溶液中に存在する各アミノ酸の数を見積る。得られた各アミノ酸の数と蛋白質のアミノ酸配列から、蛋白質濃度を決定する。しかしこの分析には、高価な装置及び高度な技術が必要であり、また委託分析にもコストと時間かかるため、現実的な方法ではない。最も実用的な手法は、Trp、Tyr、Cys残基に由来する280 nmの蛋白質溶液の吸光度を測定することである。この場合、乾燥重量法等のような他の手法により、予め正確に決定された蛋白質固有のモル吸光係数から、蛋白質濃度を決定する。モル吸光係数とは、光路長1 cmで濃度1 molの時の吸光度である(5)。蛋白質のモル吸光係数(εprotein)が未知な場合は、280 nmでのTrp、Tyr、Cys残基のモル吸光係数(εTrp=5,690、εTyr=1,280、εCys=120)を用いて、以下の式から計算できる。
\[ε_{protein} = 5,690 N_{Trp} + 1,280 N_{Tyr} +120 N_{Cys}\]NTrp、NTyr、NCysは、アミノ酸配列中のTrp、Tyr、Cys残基の数である。εproteinから以下の式を用いて、蛋白質濃度(cprotein)を求めることができる。
\[c_{protein} (mg/ml) = (分子量) x (A_{280}/ε_{protein})\]A280は、1cmあたりの280 nmにおける蛋白質溶液の吸光度である。例として、卵白リゾチームの濃度の計算方法を記す。卵白リゾチームは、1分子(分子量14,314)中に、6個のTrp、3個のTyr、8個のCysを含んでいる。この場合、εlysozyme = 5,690 x 6 + 1,280 x 3 + 120 x 8 = 38,940となり、A280が仮に0.500であれば、clysozyme = (0.500/38,940) x 14,314 = 0.184 mg/mlとなる。
STEP3 スペクトル測定の実施
・測定の準備
VUVCDとCD測定で異なる点は、VUVCDでは、1.光源が放射光であること、2.試料溶液を保持する光学セルが組み立て式であるということ、3.装置が真空対応であることが挙げられる。以下にVUVCD測定を開始するまでの手順を記す。
1.放射光の利用
放射光は、ビームラインのゲートバルブをすべて開けることで、VUVCD装置に供給される。バルブの開閉は、利用開始前にビームライン担当者が行うことがある。
2.光学セルの組み立て
VUVCD用の光学セルは、ステンレス製セルホルダー(固定ネジ、押え板、円筒部、土台部)、直径20 mm・厚さ1 mmの2枚の光学結晶板(CaF2あるいはMgF2)、ドーナツ型テフロンスペーサー、フッ素ゴム製O-ringから成る(図3)。ドーナツ型テフロンスペーサーにより、光路長が1.3~100μmの範囲で調製でき、フッ素ゴム製O-ringにより、真空環境から蛋白質溶液を密閉することができる。光路長の精度は、蛋白質濃度と同様に、スペクトルの強度に影響するため、分光光度計を用いた干渉縞の観測から正確な値を決定する。以下に光学セルの組み立て方法を記す。A.土台部に、大小2個のO-ringと1枚の光学結晶板を設置する。B.テフロンスペーサーを結晶板の上に乗せ、蛋白質溶液をマイクロピペット等で結晶板の中心にマウントする。C.蛋白質溶液とテフロンスペーサーの上にもう1枚の結晶板を置き、O-ringを取り付けた押え板を土台部の上に設置する。この時、押え板が回転しないようにストッパーで固定する。D.円筒部を土台部に取り付け、さらに固定ネジで全体を締める。ニードルの位置を確認し、締め具合を調整する。
3.真空引き
VUVCD装置の試料チャンバーと偏光変調チャンバーは高真空 (10-4Pa) に保たれ、ゲートバルブで仕切られている(図2)。蛋白質溶液を入れた光学セルを設置する際は、試料チャンバーのみを窒素ガスでリークする。設置後、真空引きを行う。
以上の作業は、測定を開始する前に、予めビームライン担当者から説明がある。2~3時間程度で、習得可能である。
・測定の開始
VUVCD測定は、測定波長範囲(nm)、走査速度(nm/min)、レスポンス(s)、データ取り込み間隔(s)、積算回数、感度(mdeg)をVUVCD測定ソフトに入力し開始する。レスポンス値を大きくし積算回数を増やすと、良好なS/N比を持ったスペクトルを得ることができる。しかし、スペクトル分解能を考慮すると、レスポンス値が大きい場合は、走査速度を遅くする必要がある。また、積算回数を増やすと、測定時間が大幅に増加する。そのため、サンプル数や放射光の利用時間を考慮してこれらの測定パラメータを決定する。また、放射光によるダメージを避けるために、同一サンプルの長時間測定はできるだけ控える。VUVCD測定ソフトは、積算されたスペクトルデータだけでなく、中間スペクトルのデータも自動的に保存するため、試料のダメージは、各中間スペクトルを比較することで確認することができる。なお、測定時間以外は、光学セルの前に設置されたシャッターにより、サンプルへの放射光を遮蔽できる(図2b)。水やほとんどの緩衝液は、CD成分を持たないが、光学結晶やCD信号処理システムにより、広い波長範囲において若干のベースラインシグナルが発生する。そのため、蛋白質溶液のスペクトルから溶媒のスペクトルを差し引くことで、真の蛋白質のCDスペクトルを得ることができる。
STEP4 スペクトル解析(二次構造解析)
・モル楕円率の計算
VUVCDスペクトルの単位は、楕円率(θ, mdeg)でVUVCD測定ソフト上に出力される。このスペクトルを濃度と光路長で規格化し、モル楕円率([θ], degrees cm2 dmol-1)あるいは、モル吸光係数(Δε)に変換する。楕円率からモル楕円率を計算するには、
\[[θ] = 100θ/Cl\]の式を使用する。l は単位がcmの光路長であり、C は単位がMのモル濃度である。また、モル吸光係数は、モル楕円率から、
\[Δε= [θ]/3,298\]の式で算出できる。蛋白質のモル楕円率を計算する場合、注意すべき点は、ここでのモル濃度は、平均残基分子量(MRW)に基づいていることである。平均残基分子量とは、蛋白質の分子量をアミノ酸残基数で割った値である。そのため、蛋白質のモル楕円率は、
\[[θ] =θx (MRW)/10cl\]から算出する方が適切である。c は単位mg/mlの濃度である。
例えば、卵白リゾチームは、分子量14,319、残基数129で、その平均残基分子量は111(14,319/129)である。実測値θが222 nmで10 (mdeg)を示し、その濃度 c が0.1 mg/ml、光路長が0.1 cmだった場合、10 x 111/(10 x 0.1 x 0.1) から、[θ]222 nmは、11,100を示す。また、その時のΔεは、11,100/3,298 から、Δε222 nm = 3.37 である。
・スペクトル解析
図4に、4種類の構造既知蛋白質(ミオグロビン、リゾチーム、コンカナバリンA、大豆トリプシンインヒビター)のVUVCDスペクトル(260~160 nm)を示す。これらのスペクトルは、モル楕円率に規格化されている。ミオグロビンは、76%のα-helix含量を持つα-helix-rich 蛋白質(α-protein)で、222と208 nm付近に負、190 nm付近に正のCDピークを持ち、さらに175 nm付近に肩、170 nm付近に負のピークを示す。また、α-helix(42%)に加えβ-strand(6%)を含んだリゾチームは、ミオグロビンと良く似たスペクトルを示すが強度が減少している。一方、46%のβ-strand含量を含むコンカナバリンAは、β-strand-rich 蛋白質(β-protein)であり、2つの負のピークが220と175nm付近に、また2つの正のピークが200と165 nm付近に存在する。しかし、同程度のβ-strand(37%)を含む大豆トリプシンインヒビターでは、220 nm付近に正、200 nm付近に大きな負のピークが存在し、β-proteinはα-proteinと比べ多様なスペクトルを示すことが分かる。またこれら構造既知蛋白質のVUVCDスペクトルから、VUV領域のCDが二次構造含量の違いを大きく反映していることが確認できる。
蛋白質二次構造には、α-helixやβ-strandに加え、turn、310helix、polyproline type II (PPII) helixが存在し、またα-helixやβ-strandは、中心付近のregular領域およびregular領域よりも歪んだ構造を持つdistorted領域(α-helixは両末端4残基、β-strandは両末端2残基)に分けられる。このdistorted領域は、α-helixとβ-strandの本数解析に利用される(6)。そのため、天然状態の構造はもとより、温度・pH・アミノ酸変異等による構造変化(非天然状態の構造)を詳細に解析するには、これら二次構造の情報を精度よく獲得する必要がある。幸いなことに、構造既知(参照)蛋白質のCDデータベースを基にした多くの二次構造解析プログラム(CONTIN、SELCON3、CDSSTR等)が開発され、実行ファイルやソースコードも公開されている。各解析法の詳細については、それぞれの文献を参照にして頂きたい(7)。
VUVCD分光法は、測定する波長領域が広範囲なため、CD分光法よりも蛋白質の二次構造の含量や本数を高い精度で算出することができ、またこれら二次構造情報を、アミノ酸配列から二次構造を予測するNeural Network(NN)法に組み込むこと(VUVCD-NN法)で、実験データを考慮した残基レベルでの二次構造の位置予測が可能である(8,9)。以下に、蛋白質の二次構造解析の手順として、31種類の参照蛋白質データベースを基にしたSELCON3プログラムによるα-helix(regularとdistorted)、β-strand(regularとdistorted)、turn、unordered構造の含量解析の方法及びα-helixとβ-strandの本数とアミノ酸配列上での位置解析の方法(A~G)を記す。また、簡単な解析ダイアグラムを図5に示す。
A.構造未知蛋白質VUVCDスペクトル(図5c)の単位をモル吸光係数(Δε)に変換し、データ間隔を1 nmに整え、SELCON3プログラムのINPUTファイルを作成する。
B.SELCON3プログラムを実行する。参照(構造既知)蛋白質のCDデータベース(図5a)と二次構造含量データベースから二次構造の成分スペクトル(図5b)が得られ、続いてINPUTファイル(図5c)が解析される。
C.SELCON3.OUTファイルから、構造未知蛋白質の6種類の二次構造の含量を確認する。α-helixとβ-strand含量は、それぞれのregularとdistorted領域の合計である。また、α-helix(あるいはβ-strand)の本数は、distorted領域の含量に未知蛋白質の残基数を掛けた後、4(あるいは2)残基で割った値である(6)。
D.PSI-BLASTプログラムにより、未知蛋白質のアミノ酸配列の位置特異的スコア行列を計算する(10)。
E. α-helixとβ-strand含量に未知蛋白質の残基数を掛けることで、α-helixとβ-strandの残基数を求める。
F.VUVCD-NNプログラムを実行する。NN法から得られた20種のアミノ酸のα-helixとβ-strandに対する重みとDからの位置特異的スコア行列、そして未知蛋白質のα-helixとβ-strandの残基数と本数情報から、二次構造の位置が計算される(8)。
G. VUVCD-NN.OUTファイルから、二次構造の位置情報を確認する(図5d)。
以上の方法から、構造未知蛋白質のVUVCDスペクトルから、二次構造含量・本数・配列(位置)を計算することができる。SELCON3プログラム、PSI-BLASTプログラム、VUVCD-NNプログラム、参照蛋白質のデータベースは、VUVCD測定用PCから利用できるため、スペクトル測定後、短時間で二次構造の解析結果を得ることができる。また、310helix、PPII の含量も計算可能である。
上述したスペクトル解析による蛋白質の構造解析の例を図6に示す。α1酸性糖蛋白質(AGP)は、天然状態ではβ-strand-rich 蛋白質であるが、生体膜存在下ではα-helix-rich 蛋白質に構造変化する(図6a)。VUVCDスペクトル解析により、AGPの天然状態と膜結合状態のα-helixやβ-strandの含量と本数が解析でき、続いてVUVCD-NN法により残基レベルでの二次構造の位置が予測できる(図6b)。このように生体膜との相互作用による二次構造変化の精密な解析結果は、蛋白質の構造-機能相関を研究する上で貴重な情報となる(11)。最近では、VUVCDからの二次構造情報は、Homology modeling等からの立体構造の予測結果を検証するデータ(図5e)としても利用される等、蛋白質構造解析におけるVUVCD分光法の用途は多方面に広がっている。
工夫とコツ
分光光度計を利用した濃度決定
分光光度計を用いた吸収測定から、蛋白質の濃度を決定する際、A280の値が0.5~1.0になるように濃度を調製し測定することが望ましい。1.0を振り切れる場合は、溶媒で希釈し測定を行う。最近では、一滴測定が可能な分光光度計のオプションが販売されており、高濃度の蛋白質の吸収を希釈無しでかつ数μlで測定できるため、これを利用しても良い。
光路長の選び方
VUV領域には水の吸収が大きいため、VUVCDスペクトルの測定限界は使用する光路長に依存する。光路長50μmでは175 nmまで、これより以下の測定には、10μmやno spacer(1.3~1.8μm)を使用する必要がある。測定限界値は、蛋白質の状態及び溶媒に使用される塩や緩衝液にも依存する。特に、no spacerの場合は、極端にCDシグナルが小さくなるため、最良の塩及び溶媒条件でかつ蛋白質濃度が高くできる時のみに使用する。
光学セルの組み立て
試料溶液上に2枚目の結晶板を置く際、気泡が形成する場合がある。これを避けるため、結晶板をピンセットでゆっくりと試料溶液上にマウントする。固定ネジのニードルの位置は、光学セルの組み立てる際、常に同じ方向を示すようにすることで、再現あるVUVCDスペクトルを得ることができる。
窒素雰囲気下での測定
VUVCD分散計では、大気の光吸収を完全に除去するため、高真空下でのCD測定が可能である。一方、蛋白質濃度や使用する溶媒の問題により、短波長側へのCD測定の拡張が十分に見込めない時等は、窒素雰囲気下の測定に変更しても良い。窒素雰囲気下では、真空ポンプを使用する必要が無くなり、通常のCD装置と同じ感覚で実験を行うことが出来る。しかし、光学セルの設置後、試料チャンバー内を十分に窒素で充満させるために15~20分程度の時間を要する。
二次構造解析で使用されるデータベース
SELCON3プログラムを用いた二次構造解析には、データベースとして31種類の構造既知蛋白質のVUVCDスペクトルと二次構造含量が使用されている。データベースは偏りの無いように、様々な二次構造成分(α-helix-richからβ-strand-richまで)を持つ蛋白質で構成されている。VUVCD-NNプログラムでは、NN法から得られた20種のアミノ酸のα-helixとβ-strandに対する重みを使用している。これらの重みは、分解能が2.0Å以下のX線結晶構造等、特定の条件を満たした計607個の蛋白質のアミノ酸配列と二次構造のデータベースから得られている(9)。二次構造解析の結果は、データベースに依存する場合がある。しかしこの性質を利用することで、解析精度を向上させることができる。例えば、VUVCDスペクトルの形状あるいはスペクトル解析の結果から、α-helix-richな蛋白質だと確認できれば、α-helix-richな蛋白質のみで構成されたデータベースをSELCON3及びNN法で使用する。この方法から、二次構造の解析精度が向上することが確認されている(9)。
実験の安全
HiSORの実験ホールへ入室するためには、被爆放射線量を測定するルクセルバッジの着用が義務付けられている。そのため、放射線業務従事状況の確認と健康診断が定期的に実施される。
文献
- Woody, R. W. /菅田宏 & 児玉高志 (訳), 蛋白質の円二色性:解析と理論に関する最近の発展 (蛋白質 核酸 酵素), 41, 56-69 (1996)
- Fasman, G. D., Circular Dichroism and the Conformational Analysis of Biomolecules, Plenum Press, New York (1996)
- Wallace, B. A. & Janes, R. W., Modern Techniques for Circular Dichroism and Synchrotron Radiation Circular Dichroism Spectroscopy, IOS, Amsterdam (2009)
- 萩原義久, 蛋白質科学会アーカイブ, 1, e002 (2008)
- Gill, S. C. & von Hippel, P. H., Anal. Biochem., 182, 319-26 (1989)
- Sreerama, N. et al., Protein Sci., 8, 370-80 (1999)
- Sreerama, N. & Woody, R. W., Anal. Biochem., 287, 252-60 (2000)
- 松尾光一, 化学と工業, 65, 227-8 (2012)
- Matsuo, K. et al., Proteins, 73, 104-12 (2008)
- Jones, D. T., J. Mol. Biol., 292, 195-202 (1999)
- Matsuo, K. et al., Biochemistry, 48, 9103-111 (2009)
-

図1 -
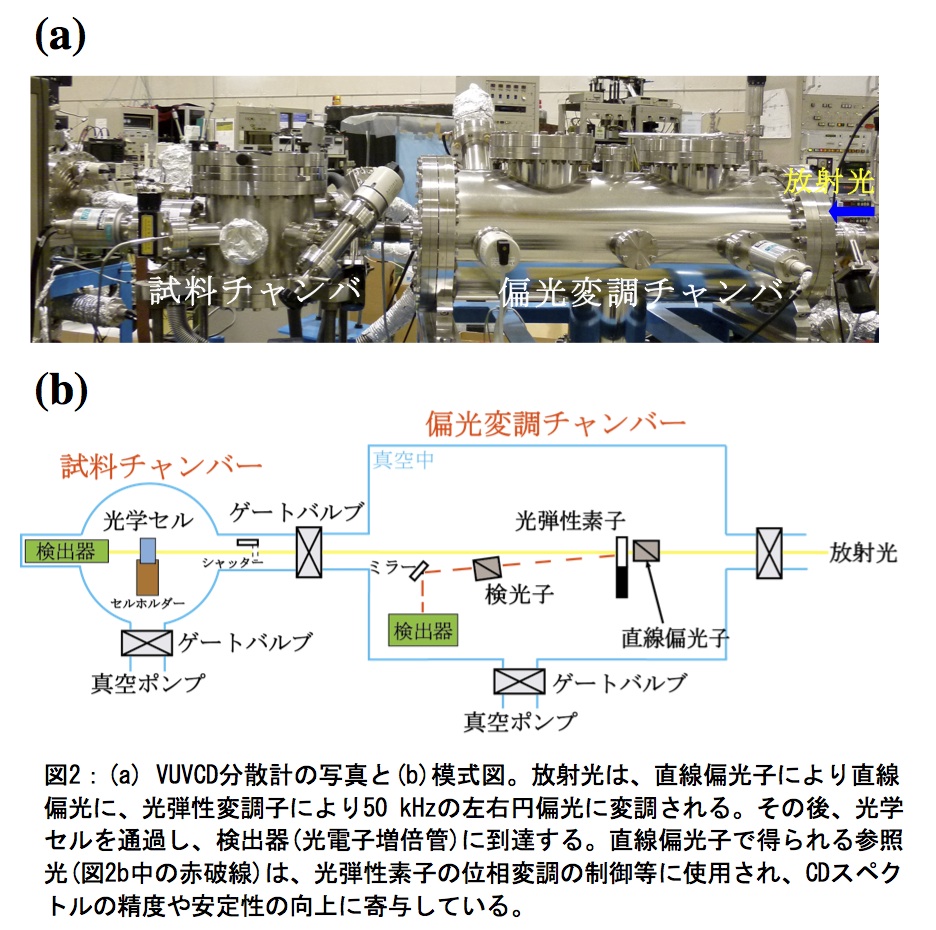
図2 -
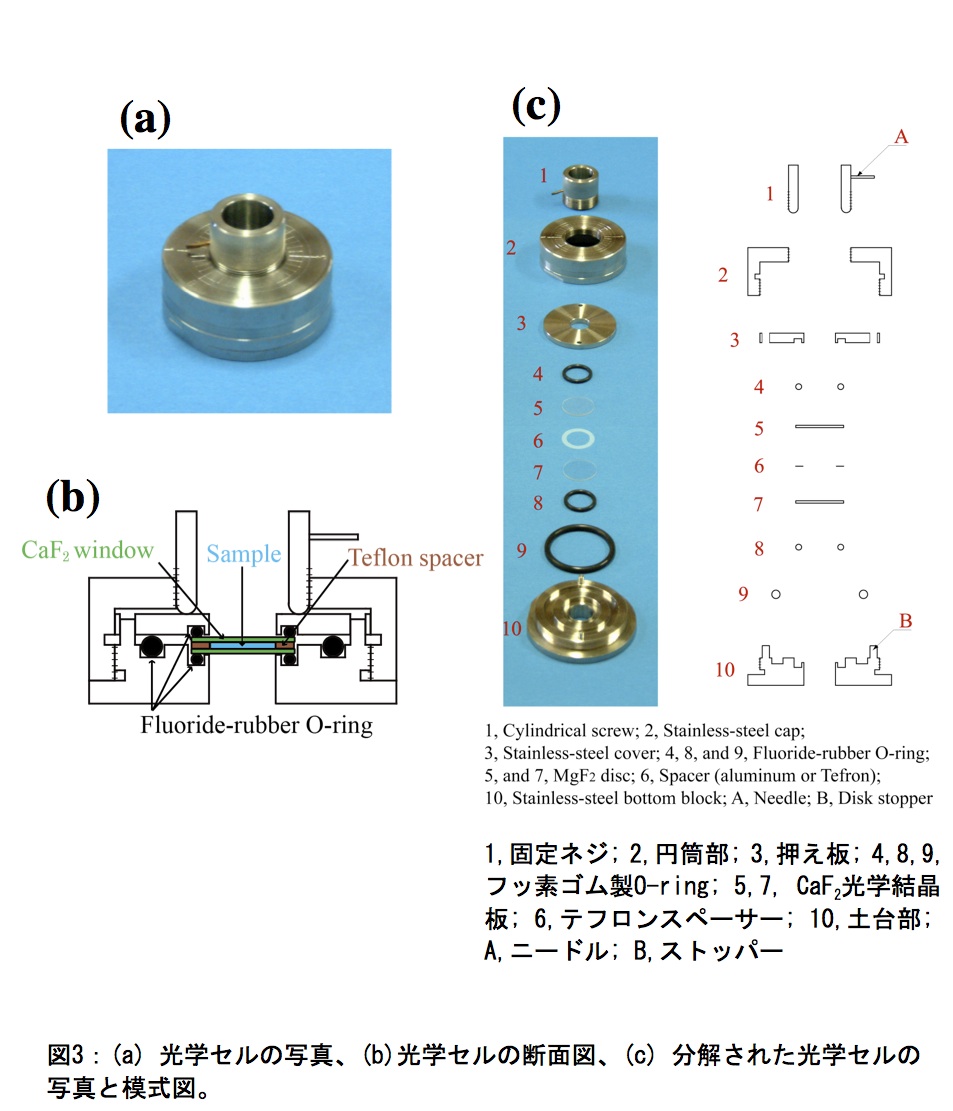
図3 -

図4 -
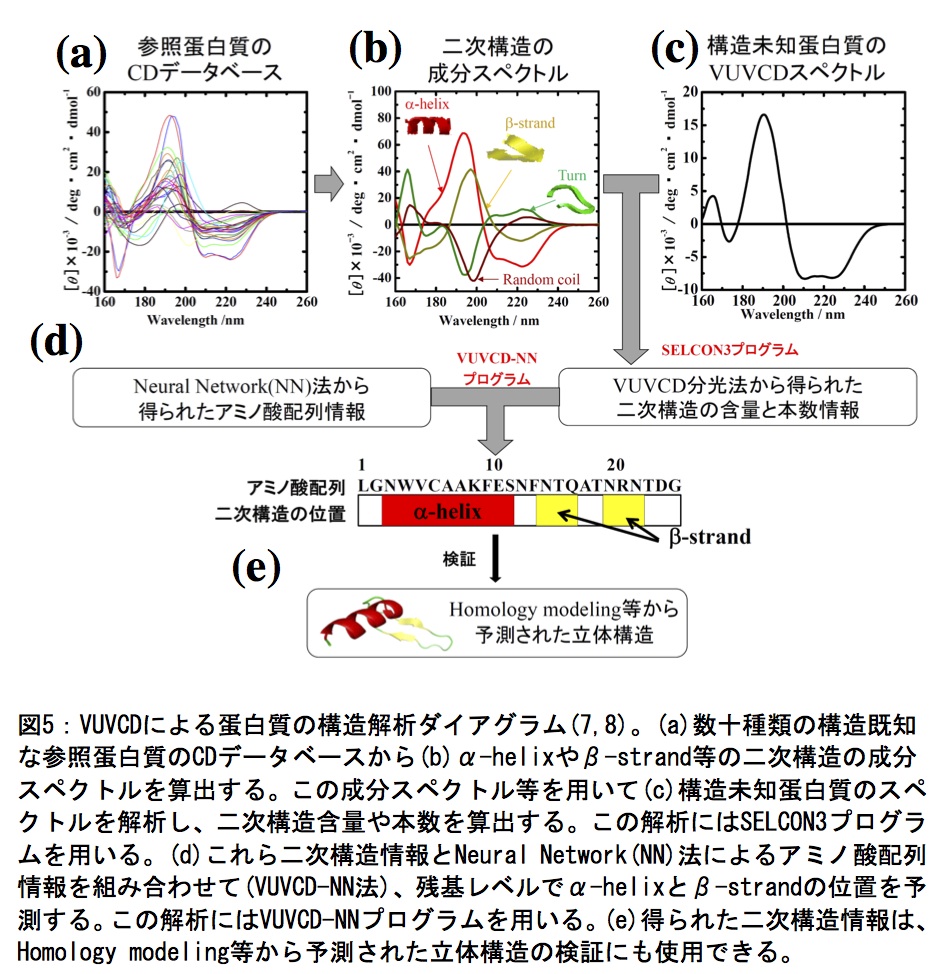
図5 -
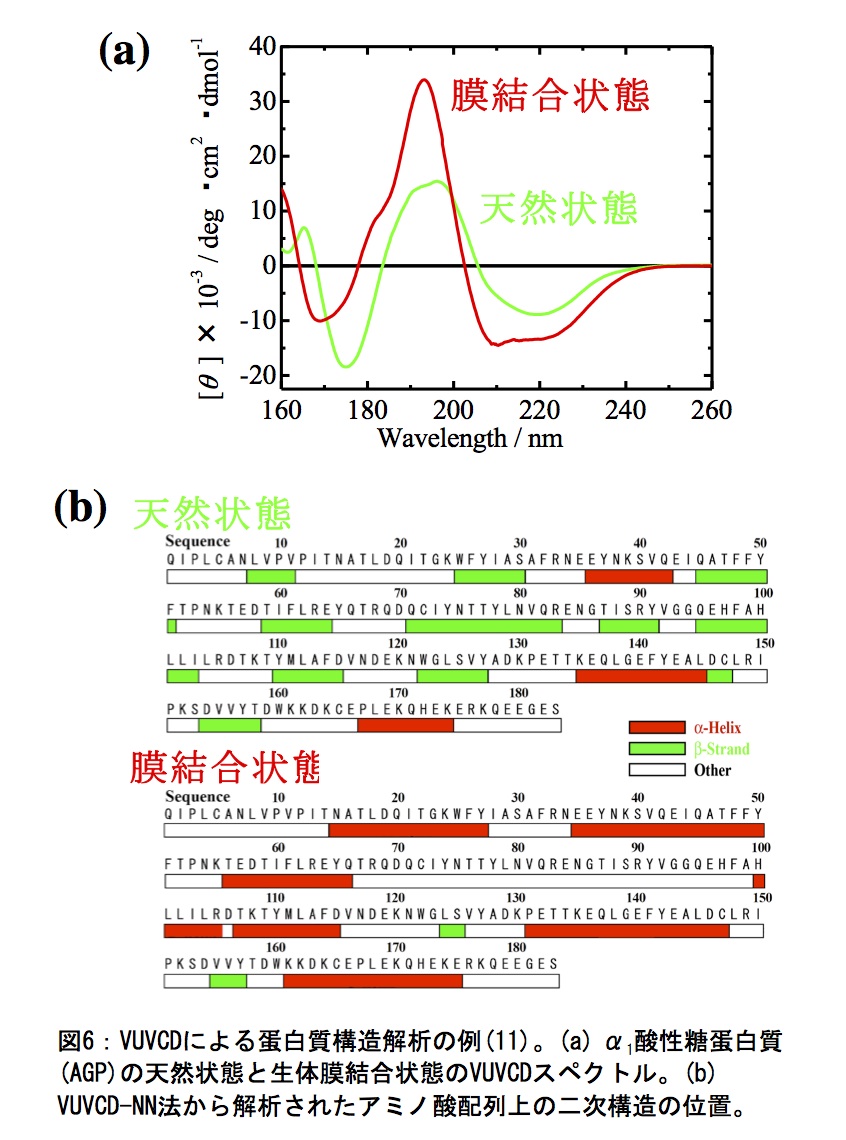
図6
概要
円二色性(CD)は、ペプチド鎖のコンフォメーションの違いを敏感に反映するため、蛋白質の二次構造の含量解析に利用されている(1,2)。またCDは、多様な実験条件下で測定できるため、X線結晶学やNMR法では難しい膜蛋白質や非天然状態の蛋白質の構造解析に有力である。しかし、通常のCD分光法は、短波長側の測定範囲が遠紫外領域である190 nm程度にとどまり、得られる二次構造情報に限界がある。真空紫外円二色性(VUVCD)分光法は、放射光を用いることで、真空紫外(VUV)領域の高エネルギー遷移に基づいたCDを検出することができ、またバイオインフォマティクス技術を取り入れたCD解析から、二次構造の含量や本数の算出に加え、アミノ酸残基レベルでの二次構造の位置予測が可能である。本稿では、広島大学放射光科学研究センター(HiSOR) のVUVCD実験のプロトコールとして、VUV領域で正確なCDスペクトルを得るための蛋白質溶液の調製及び測定方法、そして様々な二次構造情報を得るためのスペクトルの解析方法を紹介する。
イントロダクション
VUVCD測定が可能な放射光CD装置は、欧米アジアの放射光施設で10機以上開発されている(3)。我が国では、HiSOR(図1)においてCDスペクトルが140 nmまで測定できるVUVCD分散計(図2)が開発されており、現在、全国共同利用装置として稼働している。VUVCD実験を行うための手順は、基本的にはCD実験と同様であるが(4)、放射光を利用するための申請書を提出する点、またサンプルの調製や測定方法そして二次構造の解析方法で異なる点がある。以下に、実験手順を4つのSTEPに分けて説明する。
装置・器具・試薬
- 真空紫外円二色性分散計(HiSOR BL-15)
- 組み立て式光学セル(HiSOR BL-15)
- 分光光度計(例えば、JASCO V-630BIO等)
- 遠心分離機
- メンブレンフィルター(孔径: 0.2~20μm)
- 解析ソフト(OriginやMicrosoft Excel等)
- 蛋白質二次構造解析プログラム(CDPro: http://lamar.colostate.edu/~sreeram/SELCON3/ 及びDichroWeb: http://dichroweb.cryst.bbk.ac.uk/html/home.shtml を参照)
実験手順
STEP1 研究課題申請書の提出
・研究課題の申請
STEP2 サンプルの準備
・蛋白質溶液の調製
・蛋白質濃度の決定
STEP3 スペクトル測定の実施
・測定の準備
・測定の開始
STEP4 スペクトル解析(二次構造解析)
・モル楕円率の計算
・スペクトル解析
実験の詳細
STEP1 研究課題申請書の提出
・研究課題の申請
HiSORでVUVCD測定を行うには、研究内容や実験期間等を記した研究課題申請書を提出する必要がある。申請書の提出時期は、毎年1月末頃である。申請方法あるいは実験の技術的な詳細は、HiSORの担当者と相談することができる。申請書の作成から実験に至るまでの手続の流れは、HiSORのホームページ(http://www.hsrc.hiroshima-u.ac.jp/format.htm)で紹介されている。
STEP2 サンプルの準備
・蛋白質溶液の調製
スペクトルの精度は、二次構造解析の結果に影響するため、測定に適した溶液調製が必要である。蛋白質の純度は、質量分析法やSDSポリアクリルアミドゲル電気泳動等を用いて、95%以上であることを確かめる。VUVCD測定では、VUV領域における水の吸収を抑えるため、光路長が極短 (1.3~100μm)な光学セルを使用する。そのため、測定に要する蛋白質溶液は微量(1~30μl)であるが、通常のCD測定に比べ高い濃度が必要である。濃度の目安は、使用する光路長や蛋白質の種類に依存するが、50~250μM(重量パーセント濃度では、0.05~0.5%)程度が目安である。また、会合体による光散乱は、スペクトルのベースラインシフトの原因となるため、遠心分離機(例えば、20,400G・20分間)やろ過(例えば、孔径0.2~20μmのメンブレンフィルター)等で、測定前に蛋白質溶液から取り除く。
VUV領域に大きな吸収を持つ塩あるいは緩衝液は、スペクトルのS/N比の低下や測定領域の拡張の障害となるため、使用を避けるか濃度を低く抑える。塩化物イオンを含むNaCl(あるいはCaCl2)は、蛋白質の安定性・溶解性・不活性化等の理由で必須な場合を除いて使用を避けるか、イオン濃度が5 mM以下になるように抑える。塩化物イオンを含まないNaFは、150 mM程度でも吸収が少ないため、NaClの代わりに利用できる。pH 8.0~9.0の範囲の溶液調製では、NaOHの使用は避けるべきで、Tris緩衝液(< 50 mM)の利用が好ましい。Tris緩衝液を作成する時は、HClの代わりに、リン酸や硫酸を用いた方が良い。pH5.5~8.0の範囲では、リン酸緩衝液(< 50 mM)が最も適しており、Tris緩衝液や吸収の大きいHEPES緩衝液の使用は控える。また、pH5.5~3.5では、酢酸緩衝液(< 50 mM)が最も良い。pH3.5~2.0では、HClの利用が好ましく、吸収の大きいクエン酸緩衝液の使用は控える。その他の溶媒として、DTTは数 mMでは測定にほとんど影響が無いが、EDTAは0.5 mMでもスペクトルのS/N比に影響するので、できれば使用を避けたい。有機溶媒であるアセトニトリル、メタノール、エタノール、ブタノール、トリフルオロエタノール、ヘキサフルオロイソプロパノールは、VUV領域の測定に適しているが、メタノールを使用した場合、180~185 nmまでのCD測定が限界である。
以上を参考にして、塩や緩衝液の種類や濃度を決定する。可能であれば、蛋白質溶液を調製する前に、使用する溶媒のVUV領域での吸収を測定することをお勧めする。
・蛋白質濃度の決定
アミノ酸配列が既知な蛋白質の濃度を決定する最良の方法は、アミノ酸分析である。この分析では、一定量の溶液中にある蛋白質を、6 N HCl・110℃の下、24時間保持し、完全にアミノ酸単体に加水分解させる。続いて、アミノ酸分析装置により溶液中に存在する各アミノ酸の数を見積る。得られた各アミノ酸の数と蛋白質のアミノ酸配列から、蛋白質濃度を決定する。しかしこの分析には、高価な装置及び高度な技術が必要であり、また委託分析にもコストと時間かかるため、現実的な方法ではない。最も実用的な手法は、Trp、Tyr、Cys残基に由来する280 nmの蛋白質溶液の吸光度を測定することである。この場合、乾燥重量法等のような他の手法により、予め正確に決定された蛋白質固有のモル吸光係数から、蛋白質濃度を決定する。モル吸光係数とは、光路長1 cmで濃度1 molの時の吸光度である(5)。蛋白質のモル吸光係数(εprotein)が未知な場合は、280 nmでのTrp、Tyr、Cys残基のモル吸光係数(εTrp=5,690、εTyr=1,280、εCys=120)を用いて、以下の式から計算できる。
\[ε_{protein} = 5,690 N_{Trp} + 1,280 N_{Tyr} +120 N_{Cys}\]NTrp、NTyr、NCysは、アミノ酸配列中のTrp、Tyr、Cys残基の数である。εproteinから以下の式を用いて、蛋白質濃度(cprotein)を求めることができる。
\[c_{protein} (mg/ml) = (分子量) x (A_{280}/ε_{protein})\]A280は、1cmあたりの280 nmにおける蛋白質溶液の吸光度である。例として、卵白リゾチームの濃度の計算方法を記す。卵白リゾチームは、1分子(分子量14,314)中に、6個のTrp、3個のTyr、8個のCysを含んでいる。この場合、εlysozyme = 5,690 x 6 + 1,280 x 3 + 120 x 8 = 38,940となり、A280が仮に0.500であれば、clysozyme = (0.500/38,940) x 14,314 = 0.184 mg/mlとなる。
STEP3 スペクトル測定の実施
・測定の準備
VUVCDとCD測定で異なる点は、VUVCDでは、1.光源が放射光であること、2.試料溶液を保持する光学セルが組み立て式であるということ、3.装置が真空対応であることが挙げられる。以下にVUVCD測定を開始するまでの手順を記す。
1.放射光の利用
放射光は、ビームラインのゲートバルブをすべて開けることで、VUVCD装置に供給される。バルブの開閉は、利用開始前にビームライン担当者が行うことがある。
2.光学セルの組み立て
VUVCD用の光学セルは、ステンレス製セルホルダー(固定ネジ、押え板、円筒部、土台部)、直径20 mm・厚さ1 mmの2枚の光学結晶板(CaF2あるいはMgF2)、ドーナツ型テフロンスペーサー、フッ素ゴム製O-ringから成る(図3)。ドーナツ型テフロンスペーサーにより、光路長が1.3~100μmの範囲で調製でき、フッ素ゴム製O-ringにより、真空環境から蛋白質溶液を密閉することができる。光路長の精度は、蛋白質濃度と同様に、スペクトルの強度に影響するため、分光光度計を用いた干渉縞の観測から正確な値を決定する。以下に光学セルの組み立て方法を記す。A.土台部に、大小2個のO-ringと1枚の光学結晶板を設置する。B.テフロンスペーサーを結晶板の上に乗せ、蛋白質溶液をマイクロピペット等で結晶板の中心にマウントする。C.蛋白質溶液とテフロンスペーサーの上にもう1枚の結晶板を置き、O-ringを取り付けた押え板を土台部の上に設置する。この時、押え板が回転しないようにストッパーで固定する。D.円筒部を土台部に取り付け、さらに固定ネジで全体を締める。ニードルの位置を確認し、締め具合を調整する。
3.真空引き
VUVCD装置の試料チャンバーと偏光変調チャンバーは高真空 (10-4Pa) に保たれ、ゲートバルブで仕切られている(図2)。蛋白質溶液を入れた光学セルを設置する際は、試料チャンバーのみを窒素ガスでリークする。設置後、真空引きを行う。
以上の作業は、測定を開始する前に、予めビームライン担当者から説明がある。2~3時間程度で、習得可能である。
・測定の開始
VUVCD測定は、測定波長範囲(nm)、走査速度(nm/min)、レスポンス(s)、データ取り込み間隔(s)、積算回数、感度(mdeg)をVUVCD測定ソフトに入力し開始する。レスポンス値を大きくし積算回数を増やすと、良好なS/N比を持ったスペクトルを得ることができる。しかし、スペクトル分解能を考慮すると、レスポンス値が大きい場合は、走査速度を遅くする必要がある。また、積算回数を増やすと、測定時間が大幅に増加する。そのため、サンプル数や放射光の利用時間を考慮してこれらの測定パラメータを決定する。また、放射光によるダメージを避けるために、同一サンプルの長時間測定はできるだけ控える。VUVCD測定ソフトは、積算されたスペクトルデータだけでなく、中間スペクトルのデータも自動的に保存するため、試料のダメージは、各中間スペクトルを比較することで確認することができる。なお、測定時間以外は、光学セルの前に設置されたシャッターにより、サンプルへの放射光を遮蔽できる(図2b)。水やほとんどの緩衝液は、CD成分を持たないが、光学結晶やCD信号処理システムにより、広い波長範囲において若干のベースラインシグナルが発生する。そのため、蛋白質溶液のスペクトルから溶媒のスペクトルを差し引くことで、真の蛋白質のCDスペクトルを得ることができる。
STEP4 スペクトル解析(二次構造解析)
・モル楕円率の計算
VUVCDスペクトルの単位は、楕円率(θ, mdeg)でVUVCD測定ソフト上に出力される。このスペクトルを濃度と光路長で規格化し、モル楕円率([θ], degrees cm2 dmol-1)あるいは、モル吸光係数(Δε)に変換する。楕円率からモル楕円率を計算するには、
\[[θ] = 100θ/Cl\]の式を使用する。l は単位がcmの光路長であり、C は単位がMのモル濃度である。また、モル吸光係数は、モル楕円率から、
\[Δε= [θ]/3,298\]の式で算出できる。蛋白質のモル楕円率を計算する場合、注意すべき点は、ここでのモル濃度は、平均残基分子量(MRW)に基づいていることである。平均残基分子量とは、蛋白質の分子量をアミノ酸残基数で割った値である。そのため、蛋白質のモル楕円率は、
\[[θ] =θx (MRW)/10cl\]から算出する方が適切である。c は単位mg/mlの濃度である。
例えば、卵白リゾチームは、分子量14,319、残基数129で、その平均残基分子量は111(14,319/129)である。実測値θが222 nmで10 (mdeg)を示し、その濃度 c が0.1 mg/ml、光路長が0.1 cmだった場合、10 x 111/(10 x 0.1 x 0.1) から、[θ]222 nmは、11,100を示す。また、その時のΔεは、11,100/3,298 から、Δε222 nm = 3.37 である。
・スペクトル解析
図4に、4種類の構造既知蛋白質(ミオグロビン、リゾチーム、コンカナバリンA、大豆トリプシンインヒビター)のVUVCDスペクトル(260~160 nm)を示す。これらのスペクトルは、モル楕円率に規格化されている。ミオグロビンは、76%のα-helix含量を持つα-helix-rich 蛋白質(α-protein)で、222と208 nm付近に負、190 nm付近に正のCDピークを持ち、さらに175 nm付近に肩、170 nm付近に負のピークを示す。また、α-helix(42%)に加えβ-strand(6%)を含んだリゾチームは、ミオグロビンと良く似たスペクトルを示すが強度が減少している。一方、46%のβ-strand含量を含むコンカナバリンAは、β-strand-rich 蛋白質(β-protein)であり、2つの負のピークが220と175nm付近に、また2つの正のピークが200と165 nm付近に存在する。しかし、同程度のβ-strand(37%)を含む大豆トリプシンインヒビターでは、220 nm付近に正、200 nm付近に大きな負のピークが存在し、β-proteinはα-proteinと比べ多様なスペクトルを示すことが分かる。またこれら構造既知蛋白質のVUVCDスペクトルから、VUV領域のCDが二次構造含量の違いを大きく反映していることが確認できる。
蛋白質二次構造には、α-helixやβ-strandに加え、turn、310helix、polyproline type II (PPII) helixが存在し、またα-helixやβ-strandは、中心付近のregular領域およびregular領域よりも歪んだ構造を持つdistorted領域(α-helixは両末端4残基、β-strandは両末端2残基)に分けられる。このdistorted領域は、α-helixとβ-strandの本数解析に利用される(6)。そのため、天然状態の構造はもとより、温度・pH・アミノ酸変異等による構造変化(非天然状態の構造)を詳細に解析するには、これら二次構造の情報を精度よく獲得する必要がある。幸いなことに、構造既知(参照)蛋白質のCDデータベースを基にした多くの二次構造解析プログラム(CONTIN、SELCON3、CDSSTR等)が開発され、実行ファイルやソースコードも公開されている。各解析法の詳細については、それぞれの文献を参照にして頂きたい(7)。
VUVCD分光法は、測定する波長領域が広範囲なため、CD分光法よりも蛋白質の二次構造の含量や本数を高い精度で算出することができ、またこれら二次構造情報を、アミノ酸配列から二次構造を予測するNeural Network(NN)法に組み込むこと(VUVCD-NN法)で、実験データを考慮した残基レベルでの二次構造の位置予測が可能である(8,9)。以下に、蛋白質の二次構造解析の手順として、31種類の参照蛋白質データベースを基にしたSELCON3プログラムによるα-helix(regularとdistorted)、β-strand(regularとdistorted)、turn、unordered構造の含量解析の方法及びα-helixとβ-strandの本数とアミノ酸配列上での位置解析の方法(A~G)を記す。また、簡単な解析ダイアグラムを図5に示す。
A.構造未知蛋白質VUVCDスペクトル(図5c)の単位をモル吸光係数(Δε)に変換し、データ間隔を1 nmに整え、SELCON3プログラムのINPUTファイルを作成する。
B.SELCON3プログラムを実行する。参照(構造既知)蛋白質のCDデータベース(図5a)と二次構造含量データベースから二次構造の成分スペクトル(図5b)が得られ、続いてINPUTファイル(図5c)が解析される。
C.SELCON3.OUTファイルから、構造未知蛋白質の6種類の二次構造の含量を確認する。α-helixとβ-strand含量は、それぞれのregularとdistorted領域の合計である。また、α-helix(あるいはβ-strand)の本数は、distorted領域の含量に未知蛋白質の残基数を掛けた後、4(あるいは2)残基で割った値である(6)。
D.PSI-BLASTプログラムにより、未知蛋白質のアミノ酸配列の位置特異的スコア行列を計算する(10)。
E. α-helixとβ-strand含量に未知蛋白質の残基数を掛けることで、α-helixとβ-strandの残基数を求める。
F.VUVCD-NNプログラムを実行する。NN法から得られた20種のアミノ酸のα-helixとβ-strandに対する重みとDからの位置特異的スコア行列、そして未知蛋白質のα-helixとβ-strandの残基数と本数情報から、二次構造の位置が計算される(8)。
G. VUVCD-NN.OUTファイルから、二次構造の位置情報を確認する(図5d)。
以上の方法から、構造未知蛋白質のVUVCDスペクトルから、二次構造含量・本数・配列(位置)を計算することができる。SELCON3プログラム、PSI-BLASTプログラム、VUVCD-NNプログラム、参照蛋白質のデータベースは、VUVCD測定用PCから利用できるため、スペクトル測定後、短時間で二次構造の解析結果を得ることができる。また、310helix、PPII の含量も計算可能である。
上述したスペクトル解析による蛋白質の構造解析の例を図6に示す。α1酸性糖蛋白質(AGP)は、天然状態ではβ-strand-rich 蛋白質であるが、生体膜存在下ではα-helix-rich 蛋白質に構造変化する(図6a)。VUVCDスペクトル解析により、AGPの天然状態と膜結合状態のα-helixやβ-strandの含量と本数が解析でき、続いてVUVCD-NN法により残基レベルでの二次構造の位置が予測できる(図6b)。このように生体膜との相互作用による二次構造変化の精密な解析結果は、蛋白質の構造-機能相関を研究する上で貴重な情報となる(11)。最近では、VUVCDからの二次構造情報は、Homology modeling等からの立体構造の予測結果を検証するデータ(図5e)としても利用される等、蛋白質構造解析におけるVUVCD分光法の用途は多方面に広がっている。
工夫とコツ
分光光度計を利用した濃度決定
分光光度計を用いた吸収測定から、蛋白質の濃度を決定する際、A280の値が0.5~1.0になるように濃度を調製し測定することが望ましい。1.0を振り切れる場合は、溶媒で希釈し測定を行う。最近では、一滴測定が可能な分光光度計のオプションが販売されており、高濃度の蛋白質の吸収を希釈無しでかつ数μlで測定できるため、これを利用しても良い。
光路長の選び方
VUV領域には水の吸収が大きいため、VUVCDスペクトルの測定限界は使用する光路長に依存する。光路長50μmでは175 nmまで、これより以下の測定には、10μmやno spacer(1.3~1.8μm)を使用する必要がある。測定限界値は、蛋白質の状態及び溶媒に使用される塩や緩衝液にも依存する。特に、no spacerの場合は、極端にCDシグナルが小さくなるため、最良の塩及び溶媒条件でかつ蛋白質濃度が高くできる時のみに使用する。
光学セルの組み立て
試料溶液上に2枚目の結晶板を置く際、気泡が形成する場合がある。これを避けるため、結晶板をピンセットでゆっくりと試料溶液上にマウントする。固定ネジのニードルの位置は、光学セルの組み立てる際、常に同じ方向を示すようにすることで、再現あるVUVCDスペクトルを得ることができる。
窒素雰囲気下での測定
VUVCD分散計では、大気の光吸収を完全に除去するため、高真空下でのCD測定が可能である。一方、蛋白質濃度や使用する溶媒の問題により、短波長側へのCD測定の拡張が十分に見込めない時等は、窒素雰囲気下の測定に変更しても良い。窒素雰囲気下では、真空ポンプを使用する必要が無くなり、通常のCD装置と同じ感覚で実験を行うことが出来る。しかし、光学セルの設置後、試料チャンバー内を十分に窒素で充満させるために15~20分程度の時間を要する。
二次構造解析で使用されるデータベース
SELCON3プログラムを用いた二次構造解析には、データベースとして31種類の構造既知蛋白質のVUVCDスペクトルと二次構造含量が使用されている。データベースは偏りの無いように、様々な二次構造成分(α-helix-richからβ-strand-richまで)を持つ蛋白質で構成されている。VUVCD-NNプログラムでは、NN法から得られた20種のアミノ酸のα-helixとβ-strandに対する重みを使用している。これらの重みは、分解能が2.0Å以下のX線結晶構造等、特定の条件を満たした計607個の蛋白質のアミノ酸配列と二次構造のデータベースから得られている(9)。二次構造解析の結果は、データベースに依存する場合がある。しかしこの性質を利用することで、解析精度を向上させることができる。例えば、VUVCDスペクトルの形状あるいはスペクトル解析の結果から、α-helix-richな蛋白質だと確認できれば、α-helix-richな蛋白質のみで構成されたデータベースをSELCON3及びNN法で使用する。この方法から、二次構造の解析精度が向上することが確認されている(9)。
実験の安全
HiSORの実験ホールへ入室するためには、被爆放射線量を測定するルクセルバッジの着用が義務付けられている。そのため、放射線業務従事状況の確認と健康診断が定期的に実施される。
文献
- Woody, R. W. /菅田宏 & 児玉高志 (訳), 蛋白質の円二色性:解析と理論に関する最近の発展 (蛋白質 核酸 酵素), 41, 56-69 (1996)
- Fasman, G. D., Circular Dichroism and the Conformational Analysis of Biomolecules, Plenum Press, New York (1996)
- Wallace, B. A. & Janes, R. W., Modern Techniques for Circular Dichroism and Synchrotron Radiation Circular Dichroism Spectroscopy, IOS, Amsterdam (2009)
- 萩原義久, 蛋白質科学会アーカイブ, 1, e002 (2008)
- Gill, S. C. & von Hippel, P. H., Anal. Biochem., 182, 319-26 (1989)
- Sreerama, N. et al., Protein Sci., 8, 370-80 (1999)
- Sreerama, N. & Woody, R. W., Anal. Biochem., 287, 252-60 (2000)
- 松尾光一, 化学と工業, 65, 227-8 (2012)
- Matsuo, K. et al., Proteins, 73, 104-12 (2008)
- Jones, D. T., J. Mol. Biol., 292, 195-202 (1999)
- Matsuo, K. et al., Biochemistry, 48, 9103-111 (2009)
