

概要
Native mass spectrometry(native MS)は蛋白質複合体の質量を精度よく決定することができる方法である [1]。変性させたサブユニットの質量も測定すれば、複合体を構成するサブユニットの化学量論を明らかにできる。本稿では native MS の測定手順について述べる。Native MS 測定が可能な質量分析装置は Bruker 社や Thermo Scientific 社など数社から販売されているが、ここでは Waters 社の SYNAPT G2-Si(図1)を例に取り解説する。なお測定の応用例については文献 [2, 3, 4, 5] も合わせてご参考いただければと思う。実験にかかる時間は試料の純度と物性に大きく左右されるが、試料に問題がなければ1時間以内に結果を得ることが可能である。
イントロダクション
Native MS が他の質量分析法と異なる点は、水素結合や疎水性相互作用などの弱い相互作用で形成された蛋白質複合体を壊さずに質量分析する点である [1, 6, 7]。それを実現するための重要な点が三つある。一つ目は、中性 pH、生理的なイオン強度の緩衝液を用いる点である。不揮発性の塩類はエレクトロスプレーイオン化を阻害するため、基本的に揮発性の溶媒しか使えないと考えてよい。実際には、酢酸やアンモニアで pH 調整された酢酸アンモニウムバッファーがよく用いられている(図2A)。イオン強度は酢酸アンモニウムの濃度で調整される。二つ目は、穏やかなイオン化法を用いる点である。初期の研究では、Matrix Assisted Laser Desorption/Ionization(MALDI)法で native MS を行った報告もあるが、マトリックスやイオン化の為のレーザー照射の影響で複合体が解離しやすいため、現在ではエレクトロスプレーイオン化法が主にもちいられている(図2B)。三つ目は、穏やかに脱溶媒和するために、試料が通過する装置内部の真空度が急激に高くならないように制御されている点である(図2C)。一点目については、pH とイオン強度の条件検討が必要な場合があるが、あとの二点は装置があれば特に検討することはない。Native MS は、試料の純度と物性(粘度と凝集性が問題となる)さえクリアできれば、正確な化学量論が迅速に得られる便利な方法である。測定が可能な最大質量は数 MDa 程度であり [8]、18 MDa のウィルスのキャプシドが測定された例もある [9]。
装置・器具・試薬
- 質量分析装置(SYNAPT G2-Si, Waters 社)(図1)
- マイクロキャピラリー作製装置(FLAMING/BROWN MICROPIPETTE PULLER P-97, SUTTER INSTRUMENT 社)
- ガラスキャピラリー(Capillaries GC100TF-10, 外径 1.0 mm, 内径 0.78 mm, 長さ 100 mm, HARVARD APPARATUS 社)
- 金蒸着装置(Agar Sputter Coater, agar scientific 社)
- スピンカラム(MicroBiospin-6 Tris, BIO-RAD 社)
- 酢酸アンモニウム水溶液(Ammonium acetate solution for molecular biology 7.5 M, SIGMA)
- 微量高速遠心機(MX-100, TOMY 社)
- 実体顕微鏡(倍率45倍程度での観察が可能で、下から照らせる光源があるもの)
- ピンセット(0103-AA-PO, DUMONT 社, 実体顕微鏡で見て先がきちんと合っているもの)
実験手順
- 試料の準備
- マイクロキャピラリーの作製
- 試料のバッファー交換
- 測定
- マススペクトルの解析
実験の詳細
1)試料の準備
どのような試料を準備すればよいか?濃度は複合体として 10 μM 以上は必要で、できるだけ濃い方がよい。ただし凝集するとマイクロキャピラリーが詰まってスプレーされなくなるので濃縮のし過ぎには注意が必要である。量はスピンカラムにアプライできる量として 20 μL は必要である。純度はもちろん高い方がよく、SDS-PAGE で95%以上の純度はあった方がよい。重要な点は、目的蛋白質よりもイオン化しやすい夾雑物を混入させないことである。特に核酸や界面活性剤、ペプチドと思われる分子の混入が問題となることが多い。これらはゲルろ過クロマトグラフィー精製だけでは除去できないことが多いので、イオン交換クロマトグラフィー精製は行うべきである。また、その後に塩類など低分子の夾雑物を除くためにゲルろ過クロマトグラフィー精製も行った方がよい。最終的な試料のバッファー組成についてであるが、測定直前にスピンカラムで酢酸アンモニウムバッファーに置換するのでバッファー組成は基本的になんでもよい。しかし、数 M 以上の高濃度の塩、グリセロール、界面活性剤などはスピンカラムでは完全には除くことができないので避けた方がよい。
尚、ATP などの低分子を入れたまま測定することも可能であるが、S/N はかなり悪くなる。どのくらいの濃度で添加が可能かは系によって様々なので条件検討が必要である。経験的には、10 μM から 100 μM ぐらいまでは添加が可能であるが、これ以上になると S/N の低下が著しく、また添加した低分子の非特異的な結合がおきやすくなる。
2)マイクロキャピラリーの作製
試料は金でコーティングされたガラス製のマイクロキャピラリーからスプレーされる。マイクロキャピラリーの先端の内径(約数 μm)と形状はシグナルを得るための重要な要素である。作製の際にはラボグローブを着用する。ガラスキャピラリーをマイクロキャピラリー作製装置にセットし、引き延ばす。出来たマイクロキャピラリーを装置から外しガラスシャーレに入れる。金蒸着装置にマイクロキャピラリーをガラスシャーレごと入れ。蒸着を5分間行う。完成するとマイクロキャピラリーが金色になる(図3)。青く見える場合は蒸着が不十分なので、蒸着用の金を新品に交換して再度、蒸着を行う。
3)試料のバッファー交換
酢酸アンモニウムバッファーに置換することにより試料から不揮発性の塩類を除く。まず事前に目的蛋白質の等電点と質量をアミノ酸配列から計算しておく。また実験的に試料が安定な pH やイオン強度が分かっている場合には酢酸アンモニウムバッファーの pH と濃度をそれに合わせる。特に制限が無い場合には、我々は 150 mM 酢酸アンモニウムバッファー pH 6.8 で測定を行っている。バッファー交換にはスピンカラムを用いる。スピンカラムの樹脂は Tris バッファーで膨潤されているので、酢酸アンモニウムバッファーで平衡化し直す。試料 20 μL を樹脂にアプライして 1,000 G で4分間遠心し、濾液を回収する。
4)測定
試料の測定の前に装置の質量校正を行う必要がある。校正試薬として50% 2-プロパノールに溶かした 1 mg/mL ヨウ化セシウムを用いる。ヨウ化セシウムは様々な m/z を示すクラスターが高分子側まで観測されるので、蛋白質複合体の native MS 測定の質量校正にはよく用いられる。通常は m/z 50–13,000 の範囲で50点ほどデータをとって校正する。校正試薬のマススペクトルは以下に述べた試料の測定の場合と同様に取得する。
測定の際は試料をマイクロキャピラリーに注入する。前述したように、このマイクロキャピラリーの内径はシグナルを得るのに重要であり、内径の調整はマイクロキャピラリーの先端を折る長さを変えることによって行う。実際には、内径を測るのは手間なので、適当な長さのところで折って、うまくシグナルが出るように検討を行う。折るときには、実体顕微鏡下でピンセットを使い、マイクロキャピラリー先端をつまむようにして折り、先端の形状ができるだけ平らになるようにピンセットで整える。
次に、試料をマイクロキャピラリーの後ろから注入して先端に溜める。注入にはマイクロローダーという先の細いチップを使う。マイクロキャピラリーに気泡が入るとスプレーされなくなるので、できるだけ入らないように注意する。細かい気泡を除くために、先端を下にして軽く指で何回か弾いてから装置にセットする。
マイクロキャピラリーの先端が試料の吸い込み口の中央に来るように位置合わせを行う(図4)。マイクロキャピラリーの乗っているステージは前後、左右、上下に動かせるようになっているので、パソコンのディスプレイに映し出されたカメラ映像を見ながら位置を調整する。
ここでパソコン操作に移る。装置の制御と解析は MassLynx(Waters 社)という専用ソフトウェアで行う。装置の測定パラメーターを入力する(後述)。電源アイコンをクリックしてマイクロキャピラリーに電圧をかける。電圧がかかるとすぐにシグナルが出る。シグナル強度が強くなるように、スペクトルを見ながらステージの位置を調整する。さらに、スペクトルを拡大して S/N ができるだけ良くなるようにステージの位置を微調整する。装置の測定パラメーターを変えてみて S/N が良い値を探す。それから1秒間のスキャンを300回程度行うことによりデータを積算してマススペクトルを得る。
装置の測定パラメーターのうち調整することが多いパラメーターを以下に示す。数値は我々が初期値として用いている値である。
- Source
- Capillary: 1.2 kV(1 kV から1.5 kV ぐらいで検討する。弱すぎるとうまくスプレーされない。強すぎると放電してシグナルが得られない。)
- Sampling Cone: 150
- Source Offset: 150
- Collision Energy
- Trap CE: 0
- Transfer CE: 0
- Gas Control
- Trap: 5.0 mL/min(変性条件の場合は 2.0 mL/min がよい)
目的とする条件でシグナルが得られない場合には、条件検討が必要となる。まず、全くシグナルが出ない場合は、サンプルの粘度が高すぎてうまくスプレーされていないことが考えられる。この場合はサンプルを2倍、4倍と希釈していくと、ある濃度でシグナルが出はじめる。
マイクロキャピラリーの内径の調整については先に述べたが、蛋白質の種類や濃度によってきれいなスペクトルが得られる内径が異なるので、そのつど検討が必要である。また、系によっては、酢酸アンモニウムバッファーの pH やイオン強度の検討が必要な場合がある。通常は 150 mM 酢酸アンモニウムバッファー pH 6.8 で問題なくデータを取ることができるが、酢酸アンモニウムバッファーの濃度を 500 mM 程度まで上げた方が S/N の良いデータが得られる系もある。この原因としては、不揮発性の塩類の持ち込みが多い場合や蛋白質の物性によることが考えられる。
サブユニットの質量を知るために変性条件での質量分析を行う。バッファー交換したサンプルに最終濃度30%になるようにギ酸を添加して、先に述べたのと同様に測定を行う。このとき Gas Control(Trap)の値を 2 mL/min に変更すると、価数分布が広がり、解析しやすいデータが得られる。変性条件ではシャープなシグナルが得られることが多いので、積算は100回程度(約2分間)で十分である。
5)マススペクトルの解析
エレクトロスプレーイオン化の場合は通常、スペクトルは3から10本程度の多価のピークとしてあらわれる(図5A)。価数は、低 m/z から高 m/z へいくにつれて、例えば、15価、14価、13価のように1ずつ小さくなる。この価数は、エレクトロスプレーイオン化の際に蛋白質複合体に付加されたプロトンの個数を意味している。
積算したスペクトルにスムージングをかけた後、ピークの m/z を線グラフ(セントロイド)で表示させる(図5B)。質量は、隣り合うピーク(価数が \(n\) と \(n - 1\) の関係にあるもの)を指定すれば、プログラムで価数とともに自動的に計算される。変性条件における質量をサブユニット質量として同様に計算し、蛋白質複合体にどのサブユニットがいくつずつ含まれるか、化学量論を求める。
工夫とコツ
負イオンモード
蛋白質複合体の種類によってはプロトン化されにくい性質のために、正イオンモードでは観測しにくいことがある。その場合は負イオンモードで測定を行う。このとき、正イオンモードのプロトン付加とは逆に、プロトンを失った状態でイオン化される。尚、核酸も負イオンモードの方が観測しやすい。
実験の安全
マイクロキャピラリーをセットするイオン源部分(図4)には高電圧がかかるので、感電には注意する。真空が落ちた状態で高電圧がかかると、放電により装置が破損する恐れがあるので、真空の維持には注意が必要である。また、一旦、真空が落ちてしまうと、復旧までに1–2日かかる。
文献
- Hernandez, H. et al., Nat. Protocol, 2, 715–726 (2007)
- Ishii, K. et al., Biochim. Biophys. Acta., 1862, 275–286 (2018)
- Krayukhina, E. et al., mAbs, 9, 664–679 (2017)
- Wang, Q. et al., Chembiochem., 18, 2094–2098 (2017)
- Fujikawa, A. et al., Sci. Rep., 6, 20473 (2016)
- Heck, AJ., Nat. Methods, 5, 927–933 (2008)
- Leney AC., et al., J. Am. Soc. Mass Spectrom., 28, 5–13 (2017)
- Sakata E., et al., Mol. Cell., 42, 637–649 (2011)
- Snijder J., et al., Angew. Chem. Int. Ed. Engl., 52, 4020–4023 (2013)
謝辞
本稿の作成にあたり自然科学研究機構・生命創成探究センター(ExCELLS)のバイオネクストプロジェクトの支援を受けました。生命創成探究センター長の加藤晃一先生に感謝申し上げます。
-

図1:質量分析装置 -
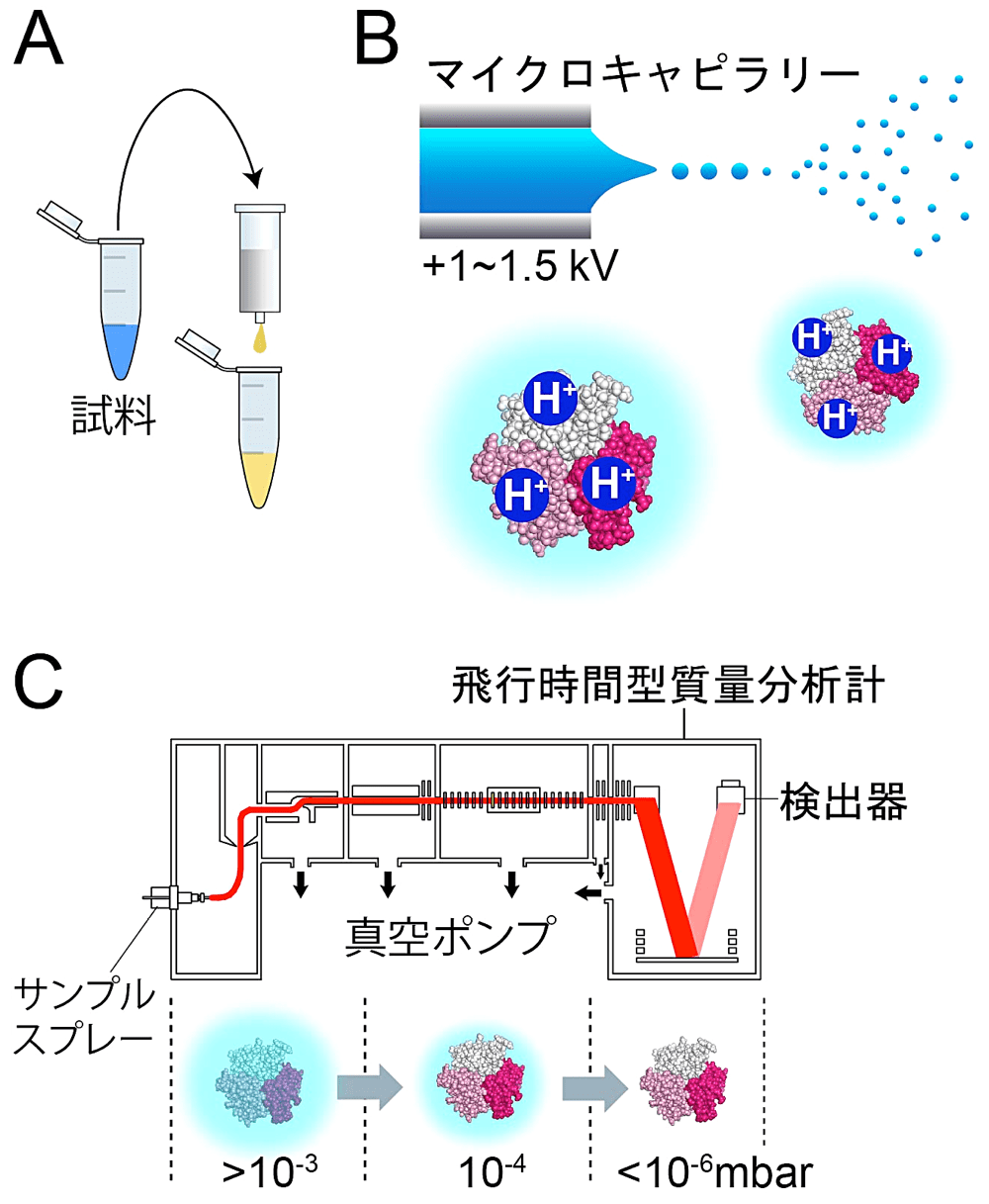
図2:Native MS の概要と装置構成。(A)バッファー交換:スピンカラムで酢酸アンモニウムバッファーに置換する。(B)エレクトロスプレーイオン化:電場により装置側へ引きつけられた液面から液滴が切り離される。液滴は細分化しつつ装置内部へと飛行し、このとき蛋白質にプロトンが付加される。(C)脱溶媒和:真空度が制御されており、蛋白質は徐々に脱溶媒和される。 -

図3:マイクロキャピラリー -

図4:(A)イオン源(B)A の白矢印方向から見た内部のコーンとマイクロキャピラリー -

図5:3量体 TNF のマススペクトル。(A)積算しスムージングをかけたマススペクトル。横軸は m/z、縦軸はシグナル強度。+15、+14、+13、+12価のピークが観測されており、質量は 55.562 Da と計算されている。(B)セントロイド。
概要
Native mass spectrometry(native MS)は蛋白質複合体の質量を精度よく決定することができる方法である [1]。変性させたサブユニットの質量も測定すれば、複合体を構成するサブユニットの化学量論を明らかにできる。本稿では native MS の測定手順について述べる。Native MS 測定が可能な質量分析装置は Bruker 社や Thermo Scientific 社など数社から販売されているが、ここでは Waters 社の SYNAPT G2-Si(図1)を例に取り解説する。なお測定の応用例については文献 [2, 3, 4, 5] も合わせてご参考いただければと思う。実験にかかる時間は試料の純度と物性に大きく左右されるが、試料に問題がなければ1時間以内に結果を得ることが可能である。
イントロダクション
Native MS が他の質量分析法と異なる点は、水素結合や疎水性相互作用などの弱い相互作用で形成された蛋白質複合体を壊さずに質量分析する点である [1, 6, 7]。それを実現するための重要な点が三つある。一つ目は、中性 pH、生理的なイオン強度の緩衝液を用いる点である。不揮発性の塩類はエレクトロスプレーイオン化を阻害するため、基本的に揮発性の溶媒しか使えないと考えてよい。実際には、酢酸やアンモニアで pH 調整された酢酸アンモニウムバッファーがよく用いられている(図2A)。イオン強度は酢酸アンモニウムの濃度で調整される。二つ目は、穏やかなイオン化法を用いる点である。初期の研究では、Matrix Assisted Laser Desorption/Ionization(MALDI)法で native MS を行った報告もあるが、マトリックスやイオン化の為のレーザー照射の影響で複合体が解離しやすいため、現在ではエレクトロスプレーイオン化法が主にもちいられている(図2B)。三つ目は、穏やかに脱溶媒和するために、試料が通過する装置内部の真空度が急激に高くならないように制御されている点である(図2C)。一点目については、pH とイオン強度の条件検討が必要な場合があるが、あとの二点は装置があれば特に検討することはない。Native MS は、試料の純度と物性(粘度と凝集性が問題となる)さえクリアできれば、正確な化学量論が迅速に得られる便利な方法である。測定が可能な最大質量は数 MDa 程度であり [8]、18 MDa のウィルスのキャプシドが測定された例もある [9]。
装置・器具・試薬
- 質量分析装置(SYNAPT G2-Si, Waters 社)(図1)
- マイクロキャピラリー作製装置(FLAMING/BROWN MICROPIPETTE PULLER P-97, SUTTER INSTRUMENT 社)
- ガラスキャピラリー(Capillaries GC100TF-10, 外径 1.0 mm, 内径 0.78 mm, 長さ 100 mm, HARVARD APPARATUS 社)
- 金蒸着装置(Agar Sputter Coater, agar scientific 社)
- スピンカラム(MicroBiospin-6 Tris, BIO-RAD 社)
- 酢酸アンモニウム水溶液(Ammonium acetate solution for molecular biology 7.5 M, SIGMA)
- 微量高速遠心機(MX-100, TOMY 社)
- 実体顕微鏡(倍率45倍程度での観察が可能で、下から照らせる光源があるもの)
- ピンセット(0103-AA-PO, DUMONT 社, 実体顕微鏡で見て先がきちんと合っているもの)
実験手順
- 試料の準備
- マイクロキャピラリーの作製
- 試料のバッファー交換
- 測定
- マススペクトルの解析
実験の詳細
1)試料の準備
どのような試料を準備すればよいか?濃度は複合体として 10 μM 以上は必要で、できるだけ濃い方がよい。ただし凝集するとマイクロキャピラリーが詰まってスプレーされなくなるので濃縮のし過ぎには注意が必要である。量はスピンカラムにアプライできる量として 20 μL は必要である。純度はもちろん高い方がよく、SDS-PAGE で95%以上の純度はあった方がよい。重要な点は、目的蛋白質よりもイオン化しやすい夾雑物を混入させないことである。特に核酸や界面活性剤、ペプチドと思われる分子の混入が問題となることが多い。これらはゲルろ過クロマトグラフィー精製だけでは除去できないことが多いので、イオン交換クロマトグラフィー精製は行うべきである。また、その後に塩類など低分子の夾雑物を除くためにゲルろ過クロマトグラフィー精製も行った方がよい。最終的な試料のバッファー組成についてであるが、測定直前にスピンカラムで酢酸アンモニウムバッファーに置換するのでバッファー組成は基本的になんでもよい。しかし、数 M 以上の高濃度の塩、グリセロール、界面活性剤などはスピンカラムでは完全には除くことができないので避けた方がよい。
尚、ATP などの低分子を入れたまま測定することも可能であるが、S/N はかなり悪くなる。どのくらいの濃度で添加が可能かは系によって様々なので条件検討が必要である。経験的には、10 μM から 100 μM ぐらいまでは添加が可能であるが、これ以上になると S/N の低下が著しく、また添加した低分子の非特異的な結合がおきやすくなる。
2)マイクロキャピラリーの作製
試料は金でコーティングされたガラス製のマイクロキャピラリーからスプレーされる。マイクロキャピラリーの先端の内径(約数 μm)と形状はシグナルを得るための重要な要素である。作製の際にはラボグローブを着用する。ガラスキャピラリーをマイクロキャピラリー作製装置にセットし、引き延ばす。出来たマイクロキャピラリーを装置から外しガラスシャーレに入れる。金蒸着装置にマイクロキャピラリーをガラスシャーレごと入れ。蒸着を5分間行う。完成するとマイクロキャピラリーが金色になる(図3)。青く見える場合は蒸着が不十分なので、蒸着用の金を新品に交換して再度、蒸着を行う。
3)試料のバッファー交換
酢酸アンモニウムバッファーに置換することにより試料から不揮発性の塩類を除く。まず事前に目的蛋白質の等電点と質量をアミノ酸配列から計算しておく。また実験的に試料が安定な pH やイオン強度が分かっている場合には酢酸アンモニウムバッファーの pH と濃度をそれに合わせる。特に制限が無い場合には、我々は 150 mM 酢酸アンモニウムバッファー pH 6.8 で測定を行っている。バッファー交換にはスピンカラムを用いる。スピンカラムの樹脂は Tris バッファーで膨潤されているので、酢酸アンモニウムバッファーで平衡化し直す。試料 20 μL を樹脂にアプライして 1,000 G で4分間遠心し、濾液を回収する。
4)測定
試料の測定の前に装置の質量校正を行う必要がある。校正試薬として50% 2-プロパノールに溶かした 1 mg/mL ヨウ化セシウムを用いる。ヨウ化セシウムは様々な m/z を示すクラスターが高分子側まで観測されるので、蛋白質複合体の native MS 測定の質量校正にはよく用いられる。通常は m/z 50–13,000 の範囲で50点ほどデータをとって校正する。校正試薬のマススペクトルは以下に述べた試料の測定の場合と同様に取得する。
測定の際は試料をマイクロキャピラリーに注入する。前述したように、このマイクロキャピラリーの内径はシグナルを得るのに重要であり、内径の調整はマイクロキャピラリーの先端を折る長さを変えることによって行う。実際には、内径を測るのは手間なので、適当な長さのところで折って、うまくシグナルが出るように検討を行う。折るときには、実体顕微鏡下でピンセットを使い、マイクロキャピラリー先端をつまむようにして折り、先端の形状ができるだけ平らになるようにピンセットで整える。
次に、試料をマイクロキャピラリーの後ろから注入して先端に溜める。注入にはマイクロローダーという先の細いチップを使う。マイクロキャピラリーに気泡が入るとスプレーされなくなるので、できるだけ入らないように注意する。細かい気泡を除くために、先端を下にして軽く指で何回か弾いてから装置にセットする。
マイクロキャピラリーの先端が試料の吸い込み口の中央に来るように位置合わせを行う(図4)。マイクロキャピラリーの乗っているステージは前後、左右、上下に動かせるようになっているので、パソコンのディスプレイに映し出されたカメラ映像を見ながら位置を調整する。
ここでパソコン操作に移る。装置の制御と解析は MassLynx(Waters 社)という専用ソフトウェアで行う。装置の測定パラメーターを入力する(後述)。電源アイコンをクリックしてマイクロキャピラリーに電圧をかける。電圧がかかるとすぐにシグナルが出る。シグナル強度が強くなるように、スペクトルを見ながらステージの位置を調整する。さらに、スペクトルを拡大して S/N ができるだけ良くなるようにステージの位置を微調整する。装置の測定パラメーターを変えてみて S/N が良い値を探す。それから1秒間のスキャンを300回程度行うことによりデータを積算してマススペクトルを得る。
装置の測定パラメーターのうち調整することが多いパラメーターを以下に示す。数値は我々が初期値として用いている値である。
- Source
- Capillary: 1.2 kV(1 kV から1.5 kV ぐらいで検討する。弱すぎるとうまくスプレーされない。強すぎると放電してシグナルが得られない。)
- Sampling Cone: 150
- Source Offset: 150
- Collision Energy
- Trap CE: 0
- Transfer CE: 0
- Gas Control
- Trap: 5.0 mL/min(変性条件の場合は 2.0 mL/min がよい)
目的とする条件でシグナルが得られない場合には、条件検討が必要となる。まず、全くシグナルが出ない場合は、サンプルの粘度が高すぎてうまくスプレーされていないことが考えられる。この場合はサンプルを2倍、4倍と希釈していくと、ある濃度でシグナルが出はじめる。
マイクロキャピラリーの内径の調整については先に述べたが、蛋白質の種類や濃度によってきれいなスペクトルが得られる内径が異なるので、そのつど検討が必要である。また、系によっては、酢酸アンモニウムバッファーの pH やイオン強度の検討が必要な場合がある。通常は 150 mM 酢酸アンモニウムバッファー pH 6.8 で問題なくデータを取ることができるが、酢酸アンモニウムバッファーの濃度を 500 mM 程度まで上げた方が S/N の良いデータが得られる系もある。この原因としては、不揮発性の塩類の持ち込みが多い場合や蛋白質の物性によることが考えられる。
サブユニットの質量を知るために変性条件での質量分析を行う。バッファー交換したサンプルに最終濃度30%になるようにギ酸を添加して、先に述べたのと同様に測定を行う。このとき Gas Control(Trap)の値を 2 mL/min に変更すると、価数分布が広がり、解析しやすいデータが得られる。変性条件ではシャープなシグナルが得られることが多いので、積算は100回程度(約2分間)で十分である。
5)マススペクトルの解析
エレクトロスプレーイオン化の場合は通常、スペクトルは3から10本程度の多価のピークとしてあらわれる(図5A)。価数は、低 m/z から高 m/z へいくにつれて、例えば、15価、14価、13価のように1ずつ小さくなる。この価数は、エレクトロスプレーイオン化の際に蛋白質複合体に付加されたプロトンの個数を意味している。
積算したスペクトルにスムージングをかけた後、ピークの m/z を線グラフ(セントロイド)で表示させる(図5B)。質量は、隣り合うピーク(価数が \(n\) と \(n - 1\) の関係にあるもの)を指定すれば、プログラムで価数とともに自動的に計算される。変性条件における質量をサブユニット質量として同様に計算し、蛋白質複合体にどのサブユニットがいくつずつ含まれるか、化学量論を求める。
工夫とコツ
負イオンモード
蛋白質複合体の種類によってはプロトン化されにくい性質のために、正イオンモードでは観測しにくいことがある。その場合は負イオンモードで測定を行う。このとき、正イオンモードのプロトン付加とは逆に、プロトンを失った状態でイオン化される。尚、核酸も負イオンモードの方が観測しやすい。
実験の安全
マイクロキャピラリーをセットするイオン源部分(図4)には高電圧がかかるので、感電には注意する。真空が落ちた状態で高電圧がかかると、放電により装置が破損する恐れがあるので、真空の維持には注意が必要である。また、一旦、真空が落ちてしまうと、復旧までに1–2日かかる。
文献
- Hernandez, H. et al., Nat. Protocol, 2, 715–726 (2007)
- Ishii, K. et al., Biochim. Biophys. Acta., 1862, 275–286 (2018)
- Krayukhina, E. et al., mAbs, 9, 664–679 (2017)
- Wang, Q. et al., Chembiochem., 18, 2094–2098 (2017)
- Fujikawa, A. et al., Sci. Rep., 6, 20473 (2016)
- Heck, AJ., Nat. Methods, 5, 927–933 (2008)
- Leney AC., et al., J. Am. Soc. Mass Spectrom., 28, 5–13 (2017)
- Sakata E., et al., Mol. Cell., 42, 637–649 (2011)
- Snijder J., et al., Angew. Chem. Int. Ed. Engl., 52, 4020–4023 (2013)
謝辞
本稿の作成にあたり自然科学研究機構・生命創成探究センター(ExCELLS)のバイオネクストプロジェクトの支援を受けました。生命創成探究センター長の加藤晃一先生に感謝申し上げます。
