

概要
膜輸送体は、脂質二重膜を介してイオン、アミノ酸、糖などの小分子を運ぶタンパク質である。一般的な酵素と異なり、膜輸送体の活性を測るためには、膜の両側を区別した反応系が必要となる。リン脂質小胞であるリポソームに膜タンパク質を再構成したプロテオリポソームは、膜内外の基質濃度、pH、溶液組成、および脂質組成、脂質:タンパク質比などを制御できることから、この目的に適している。本稿では、真核生物由来のリン酸輸送体の例を用いて、精製タンパク質および未精製膜画分をリポソームに再構成し、その活性を測る手法について紹介する。目的に応じて工夫を加えれば、様々な膜タンパク質の活性測定に応用できる。
イントロダクション
膜タンパク質は生体膜上で起こる様々な反応を担っており、その機能を理解することは、基礎研究や応用の観点から重要である。蛋白質科学会アーカイブにおいても、膜タンパク質の発現、精製、結晶化や、脂質への再構成に関する手法などが詳しく取り上げられてきた。膜タンパク質の機能を理解するうえで、その膜タンパク質が起こす反応を実際に測定することは重要であるが、一般的な酵素反応と異なり、膜上で起こる反応を測定するためには、いくつかの工夫が必要となる。特に、膜輸送体(トランスポーター)による物質輸送反応を調べるためには、膜の両側を区分けし、それらの間の基質の移動を測る必要がある。イオンチャネルの活性測定にはパッチクランプ法などの電気生理学的手法が用いられるが、速度が遅い、あるいは電荷の移動を伴わないタイプの膜輸送体にはこの手法は用いることはできない。
そのような膜輸送体の活性測定に広く用いられているのが、リポソームを用いた系である。リポソームはリン脂質二重膜からなる小胞であり、ここに目的の膜輸送体を埋め込んだプロテオリポソームは、生体膜を模した環境となる。プロテオリポソームは、適切な基質検出系(たとえば放射性標識)と組み合わせて用いれば、様々な膜輸送体の解析に利用できる。筆者らは、葉緑体由来のトリオースリン酸/リン酸輸送体(TPT)の構造に関する論文中で(文献1)、プロテオリポソームを用いた輸送活性測定を行った。この実験の元となった過去の報告(文献2)では、酵母由来の膜画分を膜融合によって直接リポソームに再構成する方法がとられていたが、筆者らはさらにアレンジを加え、精製タンパク質を用いた活性測定も行った。精製タンパク質を用いた場合、酵母膜画分を用いた場合に比べて、バックグラウンド値が低下したことによりシグナル対ノイズ比が改善された。本稿では、精製した膜タンパク質、あるいは未精製の膜画分をリポソームに再構成し、放射性標識した基質を用いて、その輸送活性を測定する実験手法について紹介したい。
装置・器具・試薬
- 装置
- 窒素ガスボンベ(各社)
- 凍結乾燥機 VD-250F(タイテック)
- 密閉式超音波破砕装置 Bioruptor UCW-310(コスモ・バイオ)
- 遠心機(2 mL チューブ用)(各社)
- 液体シンチレーションカウンタ(各社)
- 器具
- ガラスバイアル(日電理化硝子; ネジ口瓶 S-09B など)
- スピンカラム(Pierce; centrifuge columns and caps, 0.8 ml)
- 液体シンチレーション計測用ガラスバイアル(Perkin Elmer)
- 試薬
- ゲルろ過クロマトグラフィー用樹脂 Sephadex G-50 Fine(GE ヘルスケア)
- 陰イオン交換樹脂 AG1-X8(analytical grade, 200-400 mesh, acetate form)(Bio-Rad)
- 放射線標識された基質(Perkin Elmer; phosphorus-32 など)
- 固形のリン脂質(Avanti; L-α-Phosphatidylcholine, Soy-40% など)
- 液体シンチレーションカクテル Ultima Gold(Perkin Elmer)
- クロロホルム(各社)
- 各種バッファー
- 材料
- 精製した膜タンパク質
- 目的の膜タンパク質を発現させた膜画分
実験手順
- リポソームの調製
- リン脂質薄膜の調製
- 単層リポソームの調製
- プロテオリポソームの調製
- A)精製タンパク質を用いた再構成法
- B)酵母膜画分を用いた膜融合による再構成法
- リポソーム分離用の樹脂の準備
- Sephadex G-50 樹脂
- AG 1-X8 樹脂
- 放射性同位体標識した基質を用いた輸送活性測定
- プロテオリポソームの前処理
- 反応外液の作製
- 輸送反応の開始と終了
- 液体シンチレーションを用いた基質取り込み量の測定
実験の詳細
1)リポソームの調製
リン脂質薄膜の調製
- ガラスバイアルにリン脂質 500 mg をはかりとる
- 100 mg/ml となるようにクロロホルムを入れ、脂質を溶解させる
- 別のガラスバイアルに、1本当たり 50 mg の脂質となるように溶液を分注する
- 窒素ガスを吹き付け、クロロホルムを揮発させる。このとき、バイアルを横に寝かせ、回しながらガスを当てることで、内壁に脂質が固着するようにする
- バイアルを凍結乾燥器に入れてさらに一晩乾燥させ、クロロホルムを完全に除去する
- バイアルの内壁に固着したリン脂質薄膜ができる。使用時まで -80℃ に保存する
単層リポソームの調製
バッファーを調製する。このバッファーは、後にリポソーム内液となる今回の例では、対向基質の有無による輸送活性の違いを調べたいため、基質であるリン酸を含む条件と含まない条件をそれぞれ作製した。
- リン酸を含む条件:120 mM Tricine-KOH (pH 7.5), 30 mM NaH2PO4
- リン酸を含まない条件:150 mM Tricine-KOH (pH 7.5)
- リン脂質薄膜 50 mg が固着したバイアルに、バッファー 2.5 ml を加える(脂質濃度は 20 mg/ml となる)
- ボルテックスを用いて脂質を懸濁する。固形物が見えなくなるまで完全に懸濁する
- 懸濁液をエッペンチューブに移し、密閉式超音波破砕装置を用いて、4℃、10分 超音波処理する。この操作により、粒子径の均一な、単層リポソームが形成される(調製したリポソームは当日中に使うのが好ましいが、4℃ で3日程度保管しても活性に影響がないことを筆者らは確認している)
2)プロテオリポソームの調製
一般的に、プロテオリポソームの調製には高純度に精製した膜タンパク質が用いられる。精製タンパク質は界面活性剤ミセル中に可溶化されているため、リポソームへ再構成するためには界面活性剤の除去や凍結融解などの操作を加える必要がある(原理や手法に関しては文献3や文献4が詳しい)。一方で、筆者らが取り扱った TPT に関しては、タンパク質を発現させた酵母の未精製膜画分を、直接リポソームに再構成することで輸送活性を測る手法が確立されていた。この手法では、界面活性剤を用いることなく、酵母膜とリポソームを膜融合させることにより目的タンパク質を含むプロテオリポソームを調製する。本項では、A)精製タンパク質を用いた再構成法と、B)酵母膜画分を用いた膜融合による再構成法の両方について記述する。それぞれの手法のメリット、デメリットについては「工夫とコツ」を参照されたい。
A)精製タンパク質を用いた再構成法
- 目的の膜タンパク質を精製し、1~10 mg/ml 程度に濃縮する
今回の例では、昆虫細胞の組み換え発現系を用いて調製した TPT を用いた
最終的なバッファー組成:10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.01% lauryl maltose neopentyl glycol - 1)で作製したリポソーム溶液 500 μl に、脂質:タンパク質比(w/w)が100:1となるように精製タンパク質を加え、すばやく混合する
- 混合液を液体窒素に入れ瞬間凍結させたあと、室温で融解させる
- 凍結融解のサイクルを3回繰り返す。この操作により、膜タンパク質がリポソームに再構成される
- 凍結融解後のプロテオリポソームは複層リポソームとなっているため、再度 4℃、10分 超音波処理することで、粒子径の均一な、単層プロテオリポソームを形成させる
B)酵母膜画分を用いた膜融合による再構成法
- 目的の膜タンパク質を発現している膜画分をバッファーに懸濁する。今回の例では、出芽酵母 Saccharomyces cerevisiae を用いた。40 ml の培養液から膜画分を調製し、125 μl に懸濁した。バッファー組成:50 mM Tricine-KOH, pH 7.5
- 1)で作成したリポソーム溶液 475 μl に、膜画分懸濁液を19:1の割合(v/v)混合する
- 凍結融解のサイクルを3回 繰り返す。この操作により、酵母由来の膜がリポソームに融合される
- 4℃、10分 超音波処理し、粒子径の均一な、単層プロテオリポソームを形成させる
3)リポソーム分離用の樹脂の準備
反応の前後に、適切なクロマトグラフィーカラムを用いてリポソームを分離することで、外液を交換し、リポソームに取り込まれていない遊離の基質を除去する。クロマトグラフィーに用いる樹脂は膨潤および平衡化が必要なため、前日に準備する今回の例では、反応前後でそれぞれ異なる種類の樹脂を用いた。
Sephadex G-50 樹脂
ゲルろ過クロマトグラフィー樹脂。リポソーム外液を交換する目的で使用する。
- 100 ml の自立チューブに、Sephadex G-50 を 4 g はかりとる
- リポソーム外液となるバッファーを調製する(今回の例では、150 mM Tricine-KOH, pH 7.5)
- 調製したバッファー 50 ml を Sephadex G-50 に加え、強く振って撹拌する
- 室温で一晩放置し、樹脂を膨潤させる
AG 1-X8樹脂
陰イオン交換クロマトグラフィー樹脂。反応終了時、リポソーム内に取り込まれなかった遊離の基質(リン酸)を取り除く目的で使用する。
- 100 ml の自立チューブに、AG1-X8 を 25 g はかりとる
- 150 mM sodium acetate を 25 ml 加え、撹拌し、室温で一晩放置して樹脂を平衡化する
4)放射性同位体標識した基質を用いた輸送活性測定
プロテオリポソームの前処理
- 2)で作製したプロテオリポソームの外液には、30 mM PO4 が含まれているが、ゲルろ過(脱塩)にかけてこれを取り除く (これにより、外液の基質濃度を望みの値にコントロールできるようになる)
- 0.8 ml のスピンカラムを 2.0 ml フタなしチューブに取り付ける
- スピンカラムに膨潤済み Sephadex G-50 樹脂 700 μl をのせる
- 小型遠心機で 700G、1分間遠心し、溶出液を捨てる(Sephadex G-50 樹脂は柱状に固まる)
- 樹脂を崩さないように注意しつつ、スピンカラムを新しいエッペンチューブに移す
- 2)で作製したプロテオリポソーム 50 μl を樹脂の中心部にアプライする
- 小型遠心機で 700 G、1分間遠心し、溶出液を回収する(約 50 μl の外液交換済みプロテオリポソーム溶液が得られる)
以上の操作を、反応に用いるプロテオリポソーム溶液の必要量だけ行う。
反応外液の作製
放射性標識された基質を含む反応外液を作製する。
- 150 mM Tricine-KOH (pH 7.5)
- 1 mM [32P]NaH2PO4 (0.1 mCi/ml)
(放射性標識された基質を、放射性標識されていない基質と適宜混合し、望みの最終濃度に調整する)
輸送反応の開始と終了
- プロテオリポソーム 30 μl をエッペンチューブに分注し、室温に戻す
- 反応外液 30 μl を加え、反応を開始する
- 反応進行中に、AG 1-X8 カラムを用意する(スピンカラムに平衡化済み AG 1-X8 樹脂スラリー 400 μl をのせ、700 G、1分間遠心し、溶出液を捨てる。スピンカラムを新しいエッペンチューブに移す)
- 目的の反応時間が経過したら、反応液 50 μl を樹脂の中心部にアプライする
- すばやく遠心機に入れ、700 G、1分間遠心し、溶出液を回収する(約 50 μl の反応済みプロテオリポソーム溶液が得られる)
液体シンチレーションを用いた基質取り込み量の測定
- シンチレーションバイアルに液体シンチレーションカクテル 5 ml を入れる
- 液体シンチレーションカクテルに、反応済みプロテオリポソーム溶液を全量加える
- 液体シンチレーションカウンタを用いて放射線量をカウントする(必要であれば、カウント数からリン酸取り込み量を算出する)
- 複数回の実験から平均値を算出し、目的に応じて値をプロットする
工夫とコツ
様々なリポソーム調製法
リポソームの調製法には様々なバリエーションが存在する。本プロトコールでは、界面活性剤を用いずに、薄膜状に乾燥させた脂質から直接リポソームを形成させる手法を用いている。この方法は、時間を節約できるほか、界面活性剤除去時に生じるタンパク質のロスを極力防ぐことが出来る。また本プロトコールでは、界面活性剤ミセル中で精製したタンパク質を再構成する際にも界面活性剤を除く操作を行っていない。持ち込みの界面活性剤によってリポソームが溶解されるのではないかと心配されるかもしれないが、脂質濃度に対して界面活性剤濃度が十分に低ければ、リポソームは溶解されないため、少なくとも今回の例では問題はない。
基質取り込み量の経時変化の測定
本プロトコールを用いて、輸送反応を2分、5分、10分、20分、30分でそれぞれ停止し、各時点におけるカウントをプロットしたものが図1である(出典:文献1)。内液に対向基質(PO4)がある条件では経時的な[32P]PO4 の取り込みが見られるのに対して、内液に対向基質が無い条件では取り込みはほとんど見られない。このことから、目的の膜タンパク質がリン酸/リン酸のアンチポーターとして働いていることが確認できる。
ミカエリス定数の算出
本プロトコールを用いて、基質濃度を 0.3125 mM、0.625 mM、1.25 mM、2.5 mM、5 mM、10 mM にそれぞれ変え、3分時点のカウントをプロットしたものが図2である(出典:文献1)。基質濃度によってバックグラウンドの値は変わるため、目的の膜タンパク質を発現していない酵母(つまり、空ベクターを形質転換したもの)の膜画分を用いて同様の実験を行い、バックグラウンドの値を測る必要がある(図3)。この値を差し引くことで、目的の膜タンパク質による正味の輸送量が算出される。その後、非線形フィッティングなどを用いてミカエリス定数(文献5)を算出する。
競合阻害の解析
本プロトコールを用いて、基質濃度を 0.125 mM とし、競合阻害を起こすと予想される基質類似化合物を過剰量(20 mM)反応外液に加えた条件で反応を行い、3分時点のカウントをプロットしたものが図4である(出典:文献1)。ある化合物の存在下で取り込み量が減少すれば、その化合物によって阻害が起こっていることが分かる。2つ以上の基質濃度に対して、阻害化合物の濃度を振れば、阻害定数(文献6)を算出することが出来る。
プロテオリポソーム調製法のメリット、デメリット
A)精製タンパク質を用いた再構成法
「実験の詳細」で記述した通り、プロテオリポソームの調製には精製タンパク質を用いる方法が一般的である(文献3)。高純度に精製したタンパク質を用いることで、発現ホストなどに由来するバックグラウンドの影響を排除できるため、目的タンパク質の活性をより正確に調べることができる。さらに、脂質組成やタンパク質:脂質比の厳密なコントロールが可能となるため、それらの条件に依存する活性の違いも調べることもできる。一方で、タンパク質の発現・精製には費用と時間がかかるため、コストパフォーマンスは低い。例えば、変異体解析をする際には、各変異体タンパク質を mg スケールで精製する必要があるが、変異体を数十種類も調べようとなるとその手間は多大である。また、可溶化や精製が困難なターゲット(あるいはそのような変異体)に関しては、そもそも適用することができない。
B)酵母膜画分を用いた膜融合による再構成法
本プロトコールで紹介した TPT およびその類縁タンパク質に関しては、目的タンパク質を酵母の膜に発現させ、その全膜画分を用いてプロテオリポソームを調製する手法が従来用いられてきた。この手法は、精製にかかる費用と手間を省けるため、コストパフォーマンスが高い。また、界面活性剤の影響を排除できる点や、精製が困難なターゲットに関しても適用できる点などのメリットがある。ただし、この手法は発現ホストに本来備わる膜輸送系のバックグラウンドが加わるため、適用できる反応系は限られる。またタンパク質量の厳密なコントロールは出来ないため、異なるタンパク質間の活性の比較には注意が必要である。変異体解析をする際には、ウエスタンブロットなどによって野生型および変異体タンパク質の発現量を調べておく必要がある。
図5aに、未精製膜画分を再構成した場合と、精製タンパク質を再構成した場合の活性測定の結果を示した。精製タンパク質を用いた場合の方がバックグラウンドの値が低いことが分かる。
遊離基質の取り除き方
筆者らがこの実験系を立ち上げる際にもっとも重要であったのが、「反応終了時にいかに遊離基質を取り除くか」という点であった。当初は、ゲルろ過によってリポソームを分離していたが、遊離基質が除ききれず、ノイズが大きくなってしまった。そこで、過去の報告例(文献2)にならい、リン酸を強力にトラップする陰イオン交換樹脂 AG 1-X8 による分離法に変えたところ、シグナル対ノイズ比が大きく改善した(図5b)。そのため、本プロトコールを他の膜輸送体に応用する際にも、基質の特性(電荷、特定の化学基との親和性)に応じて様々な分離法を試してみるのがよい。クロマトグラフィー以外に、リポソームをフィルターに吸着させる方法もある。
基質の検出系
本プロトコールでは、放射性同位体を用いて基質を検出する方法を紹介した。この方法は、他の方法に比べて、ダイナミックレンジが広く、弱いシグナルでも検出できるメリットがある。その他にも、基質感受性の蛍光色素を用いる方法(文献7)、pH 指示薬を用いる方法(文献8)、質量分析を用いる方法(文献9)などが広く用いられている。蛍光色素や pH 指示薬を用いる場合には、リアルタイムでの計測も可能である。
実験の安全
- クロロホルムは有毒であるため、揮発させる操作はドラフト内で行うこと
- 放射性物質を扱う実験は、放射線管理区域内で行い、汚染に気を付けること
- 放射性廃棄物は所属機関の規定に従い適切に廃棄すること
文献
- Lee, Y. et al., Nat. Plants 3, 825–832 (2017)
- Linka, M. et al., Plant Physiol. 148, 1487–1496 (2008)
- Rigaud, JL. et al. Biochim. Biophys. Acta 1231, 223–246 (1995)
- Geertsma, ER. et al., Nat. Protoc. 3, 256–66 (2008)
- Johnson, KA. & Goody, RS., Biochemistry 50, 8264–8269 (2011)
- Dixon, M., Biochem. J. 55, 170–171 (1953)
- Ehrnstorfer, IA. et al., Nat. Struct. Mol. Biol. 21, 990–996 (2014)
- Lee, C. et al., Nature 501, 573–577 (2013)
- Rautengarten, C. et al., Proc. Natl. Acad. Sci. U.S.A. 111, 11563–11568 (2014)
-
![図1:時間に依存する総輸送量の変化。内液にPO4を含む条件(黒)、含まない条件(白)のそれぞれにおける[32P]PO4の取り込み量を測定した。縦軸は比活性を表す(平均値±標準誤差)。](/archives/images/memb_05/memb_05_Fig_01.png)
図1:時間に依存する総輸送量の変化。内液に PO4 を含む条件(黒)、含まない条件(白)のそれぞれにおける [32P]PO4 の取り込み量を測定した。縦軸は比活性を表す(平均値±標準誤差)。 -

図2:基質濃度に依存する輸送速度変化。縦軸は比活性を表す(平均値±標準誤差)。 -
![図3:総シグナルとバックグラウンド。輸送体を再構成したリポソーム(黒)、再構成していないリポソーム(白)それぞれへの[32P]PO4の取り込み量を測定した。縦軸は比活性を表す(平均値±標準誤差)。](/archives/images/memb_05/memb_05_Fig_03.png)
図3:総シグナルとバックグラウンド。輸送体を再構成したリポソーム(黒)、再構成していないリポソーム(白)それぞれへの [32P]PO4 の取り込み量を測定した。縦軸は比活性を表す(平均値±標準誤差)。 -

図4:競合阻害の解析。縦軸は比活性を表す(平均値±標準誤差)。 -
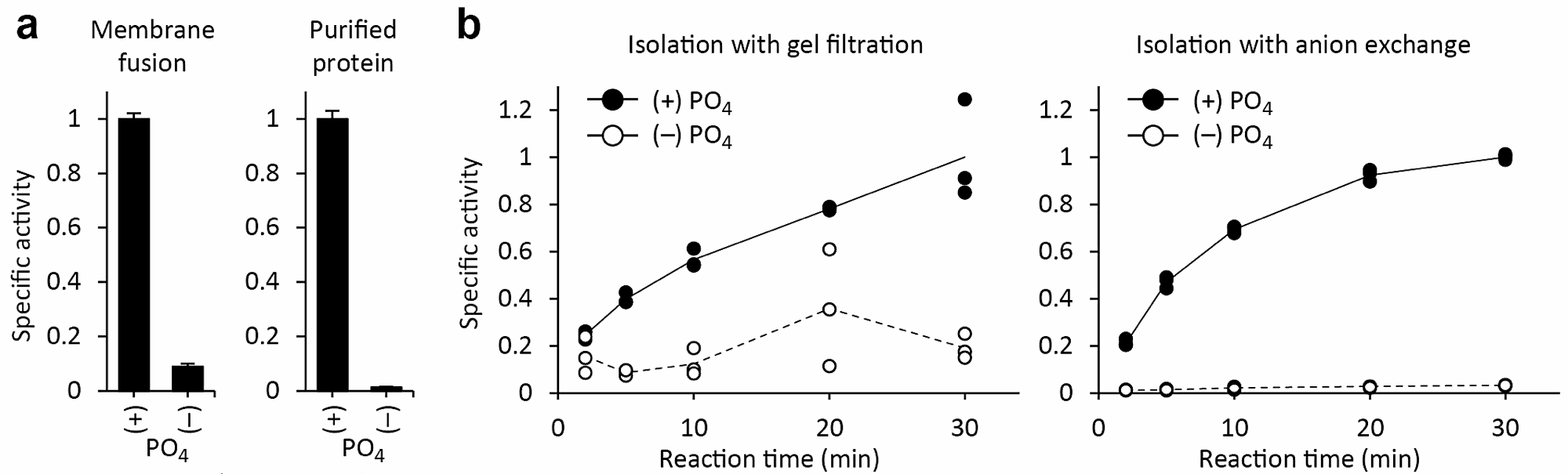
図5:実験条件の検討。a)未精製膜画分(左)、精製タンパク質(右)を用いた場合の結果を比較した。精製タンパク質を用いた方が、バックグラウンド(内液に PO4 を含まない条件下での取り込み量)が低いことが分かる。b)反応終了後のリポソームをゲルろ過(左)、陰イオン交換(右)によって単離した場合の結果を比較した。点は各試行(n = 3)におけるデータポイントを、線はそれらの平均値を示す。
概要
膜輸送体は、脂質二重膜を介してイオン、アミノ酸、糖などの小分子を運ぶタンパク質である。一般的な酵素と異なり、膜輸送体の活性を測るためには、膜の両側を区別した反応系が必要となる。リン脂質小胞であるリポソームに膜タンパク質を再構成したプロテオリポソームは、膜内外の基質濃度、pH、溶液組成、および脂質組成、脂質:タンパク質比などを制御できることから、この目的に適している。本稿では、真核生物由来のリン酸輸送体の例を用いて、精製タンパク質および未精製膜画分をリポソームに再構成し、その活性を測る手法について紹介する。目的に応じて工夫を加えれば、様々な膜タンパク質の活性測定に応用できる。
イントロダクション
膜タンパク質は生体膜上で起こる様々な反応を担っており、その機能を理解することは、基礎研究や応用の観点から重要である。蛋白質科学会アーカイブにおいても、膜タンパク質の発現、精製、結晶化や、脂質への再構成に関する手法などが詳しく取り上げられてきた。膜タンパク質の機能を理解するうえで、その膜タンパク質が起こす反応を実際に測定することは重要であるが、一般的な酵素反応と異なり、膜上で起こる反応を測定するためには、いくつかの工夫が必要となる。特に、膜輸送体(トランスポーター)による物質輸送反応を調べるためには、膜の両側を区分けし、それらの間の基質の移動を測る必要がある。イオンチャネルの活性測定にはパッチクランプ法などの電気生理学的手法が用いられるが、速度が遅い、あるいは電荷の移動を伴わないタイプの膜輸送体にはこの手法は用いることはできない。
そのような膜輸送体の活性測定に広く用いられているのが、リポソームを用いた系である。リポソームはリン脂質二重膜からなる小胞であり、ここに目的の膜輸送体を埋め込んだプロテオリポソームは、生体膜を模した環境となる。プロテオリポソームは、適切な基質検出系(たとえば放射性標識)と組み合わせて用いれば、様々な膜輸送体の解析に利用できる。筆者らは、葉緑体由来のトリオースリン酸/リン酸輸送体(TPT)の構造に関する論文中で(文献1)、プロテオリポソームを用いた輸送活性測定を行った。この実験の元となった過去の報告(文献2)では、酵母由来の膜画分を膜融合によって直接リポソームに再構成する方法がとられていたが、筆者らはさらにアレンジを加え、精製タンパク質を用いた活性測定も行った。精製タンパク質を用いた場合、酵母膜画分を用いた場合に比べて、バックグラウンド値が低下したことによりシグナル対ノイズ比が改善された。本稿では、精製した膜タンパク質、あるいは未精製の膜画分をリポソームに再構成し、放射性標識した基質を用いて、その輸送活性を測定する実験手法について紹介したい。
装置・器具・試薬
- 装置
- 窒素ガスボンベ(各社)
- 凍結乾燥機 VD-250F(タイテック)
- 密閉式超音波破砕装置 Bioruptor UCW-310(コスモ・バイオ)
- 遠心機(2 mL チューブ用)(各社)
- 液体シンチレーションカウンタ(各社)
- 器具
- ガラスバイアル(日電理化硝子; ネジ口瓶 S-09B など)
- スピンカラム(Pierce; centrifuge columns and caps, 0.8 ml)
- 液体シンチレーション計測用ガラスバイアル(Perkin Elmer)
- 試薬
- ゲルろ過クロマトグラフィー用樹脂 Sephadex G-50 Fine(GE ヘルスケア)
- 陰イオン交換樹脂 AG1-X8(analytical grade, 200-400 mesh, acetate form)(Bio-Rad)
- 放射線標識された基質(Perkin Elmer; phosphorus-32 など)
- 固形のリン脂質(Avanti; L-α-Phosphatidylcholine, Soy-40% など)
- 液体シンチレーションカクテル Ultima Gold(Perkin Elmer)
- クロロホルム(各社)
- 各種バッファー
- 材料
- 精製した膜タンパク質
- 目的の膜タンパク質を発現させた膜画分
実験手順
- リポソームの調製
- リン脂質薄膜の調製
- 単層リポソームの調製
- プロテオリポソームの調製
- A)精製タンパク質を用いた再構成法
- B)酵母膜画分を用いた膜融合による再構成法
- リポソーム分離用の樹脂の準備
- Sephadex G-50 樹脂
- AG 1-X8 樹脂
- 放射性同位体標識した基質を用いた輸送活性測定
- プロテオリポソームの前処理
- 反応外液の作製
- 輸送反応の開始と終了
- 液体シンチレーションを用いた基質取り込み量の測定
実験の詳細
1)リポソームの調製
リン脂質薄膜の調製
- ガラスバイアルにリン脂質 500 mg をはかりとる
- 100 mg/ml となるようにクロロホルムを入れ、脂質を溶解させる
- 別のガラスバイアルに、1本当たり 50 mg の脂質となるように溶液を分注する
- 窒素ガスを吹き付け、クロロホルムを揮発させる。このとき、バイアルを横に寝かせ、回しながらガスを当てることで、内壁に脂質が固着するようにする
- バイアルを凍結乾燥器に入れてさらに一晩乾燥させ、クロロホルムを完全に除去する
- バイアルの内壁に固着したリン脂質薄膜ができる。使用時まで -80℃ に保存する
単層リポソームの調製
バッファーを調製する。このバッファーは、後にリポソーム内液となる今回の例では、対向基質の有無による輸送活性の違いを調べたいため、基質であるリン酸を含む条件と含まない条件をそれぞれ作製した。
- リン酸を含む条件:120 mM Tricine-KOH (pH 7.5), 30 mM NaH2PO4
- リン酸を含まない条件:150 mM Tricine-KOH (pH 7.5)
- リン脂質薄膜 50 mg が固着したバイアルに、バッファー 2.5 ml を加える(脂質濃度は 20 mg/ml となる)
- ボルテックスを用いて脂質を懸濁する。固形物が見えなくなるまで完全に懸濁する
- 懸濁液をエッペンチューブに移し、密閉式超音波破砕装置を用いて、4℃、10分 超音波処理する。この操作により、粒子径の均一な、単層リポソームが形成される(調製したリポソームは当日中に使うのが好ましいが、4℃ で3日程度保管しても活性に影響がないことを筆者らは確認している)
2)プロテオリポソームの調製
一般的に、プロテオリポソームの調製には高純度に精製した膜タンパク質が用いられる。精製タンパク質は界面活性剤ミセル中に可溶化されているため、リポソームへ再構成するためには界面活性剤の除去や凍結融解などの操作を加える必要がある(原理や手法に関しては文献3や文献4が詳しい)。一方で、筆者らが取り扱った TPT に関しては、タンパク質を発現させた酵母の未精製膜画分を、直接リポソームに再構成することで輸送活性を測る手法が確立されていた。この手法では、界面活性剤を用いることなく、酵母膜とリポソームを膜融合させることにより目的タンパク質を含むプロテオリポソームを調製する。本項では、A)精製タンパク質を用いた再構成法と、B)酵母膜画分を用いた膜融合による再構成法の両方について記述する。それぞれの手法のメリット、デメリットについては「工夫とコツ」を参照されたい。
A)精製タンパク質を用いた再構成法
- 目的の膜タンパク質を精製し、1~10 mg/ml 程度に濃縮する
今回の例では、昆虫細胞の組み換え発現系を用いて調製した TPT を用いた
最終的なバッファー組成:10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.01% lauryl maltose neopentyl glycol - 1)で作製したリポソーム溶液 500 μl に、脂質:タンパク質比(w/w)が100:1となるように精製タンパク質を加え、すばやく混合する
- 混合液を液体窒素に入れ瞬間凍結させたあと、室温で融解させる
- 凍結融解のサイクルを3回繰り返す。この操作により、膜タンパク質がリポソームに再構成される
- 凍結融解後のプロテオリポソームは複層リポソームとなっているため、再度 4℃、10分 超音波処理することで、粒子径の均一な、単層プロテオリポソームを形成させる
B)酵母膜画分を用いた膜融合による再構成法
- 目的の膜タンパク質を発現している膜画分をバッファーに懸濁する。今回の例では、出芽酵母 Saccharomyces cerevisiae を用いた。40 ml の培養液から膜画分を調製し、125 μl に懸濁した。バッファー組成:50 mM Tricine-KOH, pH 7.5
- 1)で作成したリポソーム溶液 475 μl に、膜画分懸濁液を19:1の割合(v/v)混合する
- 凍結融解のサイクルを3回 繰り返す。この操作により、酵母由来の膜がリポソームに融合される
- 4℃、10分 超音波処理し、粒子径の均一な、単層プロテオリポソームを形成させる
3)リポソーム分離用の樹脂の準備
反応の前後に、適切なクロマトグラフィーカラムを用いてリポソームを分離することで、外液を交換し、リポソームに取り込まれていない遊離の基質を除去する。クロマトグラフィーに用いる樹脂は膨潤および平衡化が必要なため、前日に準備する今回の例では、反応前後でそれぞれ異なる種類の樹脂を用いた。
Sephadex G-50 樹脂
ゲルろ過クロマトグラフィー樹脂。リポソーム外液を交換する目的で使用する。
- 100 ml の自立チューブに、Sephadex G-50 を 4 g はかりとる
- リポソーム外液となるバッファーを調製する(今回の例では、150 mM Tricine-KOH, pH 7.5)
- 調製したバッファー 50 ml を Sephadex G-50 に加え、強く振って撹拌する
- 室温で一晩放置し、樹脂を膨潤させる
AG 1-X8樹脂
陰イオン交換クロマトグラフィー樹脂。反応終了時、リポソーム内に取り込まれなかった遊離の基質(リン酸)を取り除く目的で使用する。
- 100 ml の自立チューブに、AG1-X8 を 25 g はかりとる
- 150 mM sodium acetate を 25 ml 加え、撹拌し、室温で一晩放置して樹脂を平衡化する
4)放射性同位体標識した基質を用いた輸送活性測定
プロテオリポソームの前処理
- 2)で作製したプロテオリポソームの外液には、30 mM PO4 が含まれているが、ゲルろ過(脱塩)にかけてこれを取り除く (これにより、外液の基質濃度を望みの値にコントロールできるようになる)
- 0.8 ml のスピンカラムを 2.0 ml フタなしチューブに取り付ける
- スピンカラムに膨潤済み Sephadex G-50 樹脂 700 μl をのせる
- 小型遠心機で 700G、1分間遠心し、溶出液を捨てる(Sephadex G-50 樹脂は柱状に固まる)
- 樹脂を崩さないように注意しつつ、スピンカラムを新しいエッペンチューブに移す
- 2)で作製したプロテオリポソーム 50 μl を樹脂の中心部にアプライする
- 小型遠心機で 700 G、1分間遠心し、溶出液を回収する(約 50 μl の外液交換済みプロテオリポソーム溶液が得られる)
以上の操作を、反応に用いるプロテオリポソーム溶液の必要量だけ行う。
反応外液の作製
放射性標識された基質を含む反応外液を作製する。
- 150 mM Tricine-KOH (pH 7.5)
- 1 mM [32P]NaH2PO4 (0.1 mCi/ml)
(放射性標識された基質を、放射性標識されていない基質と適宜混合し、望みの最終濃度に調整する)
輸送反応の開始と終了
- プロテオリポソーム 30 μl をエッペンチューブに分注し、室温に戻す
- 反応外液 30 μl を加え、反応を開始する
- 反応進行中に、AG 1-X8 カラムを用意する(スピンカラムに平衡化済み AG 1-X8 樹脂スラリー 400 μl をのせ、700 G、1分間遠心し、溶出液を捨てる。スピンカラムを新しいエッペンチューブに移す)
- 目的の反応時間が経過したら、反応液 50 μl を樹脂の中心部にアプライする
- すばやく遠心機に入れ、700 G、1分間遠心し、溶出液を回収する(約 50 μl の反応済みプロテオリポソーム溶液が得られる)
液体シンチレーションを用いた基質取り込み量の測定
- シンチレーションバイアルに液体シンチレーションカクテル 5 ml を入れる
- 液体シンチレーションカクテルに、反応済みプロテオリポソーム溶液を全量加える
- 液体シンチレーションカウンタを用いて放射線量をカウントする(必要であれば、カウント数からリン酸取り込み量を算出する)
- 複数回の実験から平均値を算出し、目的に応じて値をプロットする
工夫とコツ
様々なリポソーム調製法
リポソームの調製法には様々なバリエーションが存在する。本プロトコールでは、界面活性剤を用いずに、薄膜状に乾燥させた脂質から直接リポソームを形成させる手法を用いている。この方法は、時間を節約できるほか、界面活性剤除去時に生じるタンパク質のロスを極力防ぐことが出来る。また本プロトコールでは、界面活性剤ミセル中で精製したタンパク質を再構成する際にも界面活性剤を除く操作を行っていない。持ち込みの界面活性剤によってリポソームが溶解されるのではないかと心配されるかもしれないが、脂質濃度に対して界面活性剤濃度が十分に低ければ、リポソームは溶解されないため、少なくとも今回の例では問題はない。
基質取り込み量の経時変化の測定
本プロトコールを用いて、輸送反応を2分、5分、10分、20分、30分でそれぞれ停止し、各時点におけるカウントをプロットしたものが図1である(出典:文献1)。内液に対向基質(PO4)がある条件では経時的な[32P]PO4 の取り込みが見られるのに対して、内液に対向基質が無い条件では取り込みはほとんど見られない。このことから、目的の膜タンパク質がリン酸/リン酸のアンチポーターとして働いていることが確認できる。
ミカエリス定数の算出
本プロトコールを用いて、基質濃度を 0.3125 mM、0.625 mM、1.25 mM、2.5 mM、5 mM、10 mM にそれぞれ変え、3分時点のカウントをプロットしたものが図2である(出典:文献1)。基質濃度によってバックグラウンドの値は変わるため、目的の膜タンパク質を発現していない酵母(つまり、空ベクターを形質転換したもの)の膜画分を用いて同様の実験を行い、バックグラウンドの値を測る必要がある(図3)。この値を差し引くことで、目的の膜タンパク質による正味の輸送量が算出される。その後、非線形フィッティングなどを用いてミカエリス定数(文献5)を算出する。
競合阻害の解析
本プロトコールを用いて、基質濃度を 0.125 mM とし、競合阻害を起こすと予想される基質類似化合物を過剰量(20 mM)反応外液に加えた条件で反応を行い、3分時点のカウントをプロットしたものが図4である(出典:文献1)。ある化合物の存在下で取り込み量が減少すれば、その化合物によって阻害が起こっていることが分かる。2つ以上の基質濃度に対して、阻害化合物の濃度を振れば、阻害定数(文献6)を算出することが出来る。
プロテオリポソーム調製法のメリット、デメリット
A)精製タンパク質を用いた再構成法
「実験の詳細」で記述した通り、プロテオリポソームの調製には精製タンパク質を用いる方法が一般的である(文献3)。高純度に精製したタンパク質を用いることで、発現ホストなどに由来するバックグラウンドの影響を排除できるため、目的タンパク質の活性をより正確に調べることができる。さらに、脂質組成やタンパク質:脂質比の厳密なコントロールが可能となるため、それらの条件に依存する活性の違いも調べることもできる。一方で、タンパク質の発現・精製には費用と時間がかかるため、コストパフォーマンスは低い。例えば、変異体解析をする際には、各変異体タンパク質を mg スケールで精製する必要があるが、変異体を数十種類も調べようとなるとその手間は多大である。また、可溶化や精製が困難なターゲット(あるいはそのような変異体)に関しては、そもそも適用することができない。
B)酵母膜画分を用いた膜融合による再構成法
本プロトコールで紹介した TPT およびその類縁タンパク質に関しては、目的タンパク質を酵母の膜に発現させ、その全膜画分を用いてプロテオリポソームを調製する手法が従来用いられてきた。この手法は、精製にかかる費用と手間を省けるため、コストパフォーマンスが高い。また、界面活性剤の影響を排除できる点や、精製が困難なターゲットに関しても適用できる点などのメリットがある。ただし、この手法は発現ホストに本来備わる膜輸送系のバックグラウンドが加わるため、適用できる反応系は限られる。またタンパク質量の厳密なコントロールは出来ないため、異なるタンパク質間の活性の比較には注意が必要である。変異体解析をする際には、ウエスタンブロットなどによって野生型および変異体タンパク質の発現量を調べておく必要がある。
図5aに、未精製膜画分を再構成した場合と、精製タンパク質を再構成した場合の活性測定の結果を示した。精製タンパク質を用いた場合の方がバックグラウンドの値が低いことが分かる。
遊離基質の取り除き方
筆者らがこの実験系を立ち上げる際にもっとも重要であったのが、「反応終了時にいかに遊離基質を取り除くか」という点であった。当初は、ゲルろ過によってリポソームを分離していたが、遊離基質が除ききれず、ノイズが大きくなってしまった。そこで、過去の報告例(文献2)にならい、リン酸を強力にトラップする陰イオン交換樹脂 AG 1-X8 による分離法に変えたところ、シグナル対ノイズ比が大きく改善した(図5b)。そのため、本プロトコールを他の膜輸送体に応用する際にも、基質の特性(電荷、特定の化学基との親和性)に応じて様々な分離法を試してみるのがよい。クロマトグラフィー以外に、リポソームをフィルターに吸着させる方法もある。
基質の検出系
本プロトコールでは、放射性同位体を用いて基質を検出する方法を紹介した。この方法は、他の方法に比べて、ダイナミックレンジが広く、弱いシグナルでも検出できるメリットがある。その他にも、基質感受性の蛍光色素を用いる方法(文献7)、pH 指示薬を用いる方法(文献8)、質量分析を用いる方法(文献9)などが広く用いられている。蛍光色素や pH 指示薬を用いる場合には、リアルタイムでの計測も可能である。
実験の安全
- クロロホルムは有毒であるため、揮発させる操作はドラフト内で行うこと
- 放射性物質を扱う実験は、放射線管理区域内で行い、汚染に気を付けること
- 放射性廃棄物は所属機関の規定に従い適切に廃棄すること
文献
- Lee, Y. et al., Nat. Plants 3, 825–832 (2017)
- Linka, M. et al., Plant Physiol. 148, 1487–1496 (2008)
- Rigaud, JL. et al. Biochim. Biophys. Acta 1231, 223–246 (1995)
- Geertsma, ER. et al., Nat. Protoc. 3, 256–66 (2008)
- Johnson, KA. & Goody, RS., Biochemistry 50, 8264–8269 (2011)
- Dixon, M., Biochem. J. 55, 170–171 (1953)
- Ehrnstorfer, IA. et al., Nat. Struct. Mol. Biol. 21, 990–996 (2014)
- Lee, C. et al., Nature 501, 573–577 (2013)
- Rautengarten, C. et al., Proc. Natl. Acad. Sci. U.S.A. 111, 11563–11568 (2014)
