

概要
機能未知のタンパク質を解析する際、相互作用するタンパク質を調べることは最も重要な解析方法の一つである。実際に相互作用するタンパク質を単離、同定するためには、目的とするタンパク質に対する特異性の高い抗体を作成し、その抗体を用いた免疫沈降法を行うことが古くから行われてきた。しかし、抗体作成にかかる時間や労力、さらには常に免疫沈降に適した抗体が入手できるわけではないという理由から、現在では「アフィニティータグ」と呼ばれる、抗原としての有用性が確認されている短いポリペプチドを目的のタンパク質のN末端やC末端に融合し、このタグを用いて相互作用因子を解析する方法が一般的となっている。これまでに様々なアフィニティータグが開発され解析に用いられているが、どのタグにもメリットとデメリットが存在し、実験の目的に応じて選択する必要がある(1)。本稿では、種々のアフィニティータグの中からタンパク質複合体の解析を目的とした効率の良いアフィニティー精製手法について述べる。
イントロダクション
本稿で述べる実験の目的は、タンパク質複合体の解析のための精製方法である。一つのアフィニティータグを用いてタンパク質複合体を精製した場合、その精製分画に含まれるペプチドは質量分析によって同定が可能である。しかし、例えば目的のタンパク質が複数の異なる複合体を形成しているような場合、どのペプチドがどの複合体に含まれているかなど、複合体の性状を詳しく解析する必要がある。実際、一度アフィニティー精製した複合体をさらに、ゲルろ過クロマトグラフィー、密度勾配遠心、二次元電気泳動などで分離し、それぞれの複合体の構成因子、酵素活性などを解析する例がいくつか報告されている。しかしながら、このような解析を行っているこれまでの多くの報告では、一回の精製で使われる生体試料として、浮遊細胞なら何十リットル、接着細胞なら大きいシャーレ何百枚という量に相当する細胞の抽出液を用いている。このような実験は費用、時間、設備など、実施するためのハードルが高い。そこでより精製効率が高い精製システムを使用すれば、このような複合体解析が容易に行えるようになると期待される。
複合体を解析するための精製手法には以下の条件が要求される:
- 精製度(特異性)が高いこと
- 収量が多いこと
- 競合反応による溶出が容易なこと
- 反応条件が穏やかであること
精製度を上げるために使われる手法としてTAP(Tandem Affinity Purification)法がよく知られている(2)。オリジナルのTAPタグは酵母のタンパク質精製でよく使用されるが、そのタグの大きさや精製効率の低さからそのまま哺乳類で使用されている例は少ない。ヒト培養細胞からの精製で頻繁に使用されるタグは、FLAGタグやHAタグなどのエピトープタグである。特にFLAGタグは特異性が高く、操作も簡便なので広く使われている。しかし条件によっては非特異的な精製産物が生じるので、精製度を上げるためにこれらエピトープタグやその他のタグをタンデムにつなげて、2段階で精製する手法もよく使われている。当研究室でもFLAGタグを使用して、質量分析による相互作用因子の同定を行っていたが、その後でさらに複合体を解析するための十分な精製産物を得るのが難しかった。そこで以下の2つのタグについてその精製効率を検証することにした。
- 3xFLAGタグ:一般的なFLAGタグ(DYKDDDK)の中で抗原性に主に寄与する部分配列をタンデムにつなげたタグ。単独のFLAGタグに比べ特異性が大きく上昇している。主に検出感度を上げるツールとして推奨されているが、精製への応用例やプロトコールはまだ多くない。
アミノ酸配列 DYKDHDGDYKDHDIDYKDDDK - SBPタグ:ストレプトアビジン–ビオチンの強固な相互作用を利用した精製手法である。原理的にはストレプタグを改良したタグであり、ストレプトアビジンをより特異的に認識することができる(3)。比較的新しいタグなので精製に利用されている実際のデータが少ない。
アミノ酸配列 MDEKTTGWRGGHVVEGLAGELEQLRARLEHHPQGQREP
これら二つのタグの利点として特異性が高いこと、競合反応による溶出が可能なこと、反応はすべて低温条件下で可能であり、精製条件が穏やかなことなどが挙げられる。ただし、3xFLAGタグ、SBPタグは共に精製するためのツールとしての報告が少なく、実際どの程度の精製効率で精製産物が得られるのか明らかにされていない。そこで本実験ではこれらのタグの比較検討を行うことにした。動物細胞の核抽出液をスタート材料として比較検討を行った結果、どちらのタグも単独の精製で非常に高い効率で精製産物が得られる事が分かった(図4、図5)。また、両者をタンデムにしたTAP法では、精製特異性が上がるものの、最終的な効率は逆に減少することが明らかになった。本プロトコールでは、ヒト培養細胞を用いたタンパク質精製までの一連の流れについて、ヒトMRG15複合体をモデルとして実際の精製過程を示すことにする(4, 5)。
装置・器具・試薬
- 冷却遠心機(各社)
- ローテーター(各社)
- ANTI-FLAG M2 Affinity Gel(Sigma #A2220)
- 3xFLAG peptide(Sigma #F4799)
- Streptavidin Sephsrose High Performance(Cytiva #17-5113-01)
- Sepharose 4B(Cytiva#17-0120-01)
- Biotin(各社、例:Fujifilm Wako #029-08713)
- 哺乳類発現用ベクター(各社、例:pcDNA4/TO,Invitrogen #V1020-20)
- リポフェクション試薬(各社、例:PolyFect Transfection Reagent,QIAGEN #301105)
- プロテアーゼインヒビター(各社、例:Complete, Roche #1873580)
- IPバッファー:
0.25 M KCl,50 mM HEPES(pH 7.9),10% glycerol, 0.2 mM EDTA,0.1% TritonX-100 - 溶出バッファー:
3xFLAGタグ:250 µg/mL 3xFLAG peptide
SBPタグ:2 mM biotin
それぞれプロテアーゼインヒビター入りのIPバッファーで希釈し、使用する。
実験手順
- タグ融合タンパク質発現細胞株の単離まで(1ヶ月〜数ヶ月)
- タグ融合タンパク質発現コンストラクトの作製(数日〜)
- 安定発現細胞株の単離(数週間〜)
- 細胞の回収(数週間)
- タンパク質画分の粗精製
- 核抽出液の調製(1日)
- アフィニティー精製(1〜2日)
- プレクリア(前処理)(30分)
- アフィニティーレジンとの結合(1時間〜O/N)
- 洗浄
- 溶出(数時間〜)
- 精製産物の確認
- ウェスタンブロット
- SDS-PAGE(銀染色など)
実験の詳細
1. タグ融合タンパク質発現細胞株の単離まで
外来タンパク質を発現させるには、トランスフェクションしたプラスミドから一過的に発現させる系と、ゲノムに導入されたコンストラクトから安定に発現させる系がある。今回のような複合体解析を目的とした実験には、比較的多くの細胞からタンパク質を調製し精製に用いる必要があり、かつ均一なサンプルを用いる方が望ましい。したがって、時間と労力はかかるものの、安定発現細胞株を単離して用いることを推奨する。
1-1. タグ融合タンパク質発現コンストラクトの作製
市販のpcDNA4TO(Invitrogen)の選択マーカーをピューロマイシン耐性遺伝子に置換したpcDNA4/TO/puroのMCS上流に、2種類の合成オリゴDNAを用いて調製した3xFLAGに相当するカセットを挿入し3xFLAG融合タンパク質発現ベクターpcDNA4/TO/3xFLAG/puroを作製した(図1、図2)。さらに、市販のpNTAP(Stratagene)からPCRで増幅させたSBP融合領域を3xFLAGコード領域の下流に挿入し、pcDNA4/TO/3xFLAG-SBP/puroを作製した(図1、図3)。それぞれのベクターに目的のタンパク質(ヒトMRG15)のcDNAをクローニングした1。
1-2. 安定発現細胞株の単離
コンストラクトが完成したら、適当な制限酵素部位を利用してプラスミドを一本鎖化し、ポリフェクト等の試薬を用いて細胞にトランスフェクションする2。35 mmのディッシュにトランスフェクションし、2日ほど培養した後、1/10程度の細胞を10 cmのディッシュに撒き直し、プラスミド上の選択マーカーによるセレクションを行う。培地の交換を2、3日おきに行い、単一な細胞からコロニーを形成するまで培養を続ける。クローニングリング(クローニングシリンダー)等を使って、単一クローンを分離し、細胞を増やしながらタグ融合タンパク質の発現の有無(誘導発現するかどうか、発現している細胞の割合等)をウェスタンブロット等でチェックし、発現が確認された細胞株をストックする。過剰発現による非生理的な影響を除くため、誘導発現条件下で内在性のタンパク質と同程度(あるいはそれ以下)の発現レベルの細胞株をタンパク質精製に使用するのが望ましい。
1-3. 細胞の回収
必要量の細胞を培養し3、接着細胞ならばPBSで洗浄した後、スクレーパーで掻き集めて、15 mLチューブに回収する。2,000 rpmで10分間遠心してペレットにし、目的に応じてタンパク質画分を粗精製する。(以下参照)
2. タンパク質画分の粗精製
精製は全細胞抽出液からでも可能である。しかしながら、膜タンパク質など不溶性画分にタンパク質が含まれている場合、可溶化してタンパク質画分を調製する必要がある。また、目的のタンパク質が核タンパク質である場合、核抽出液を調製してから精製を行うことが一般的であり、より高い精製度を得ることができる。核抽出液調製法はDignam法が広く利用されている。バッファーの組成、細かい点などは原著論文(6)やタンパク質実験のプロトコール集などを参考にしてもらいたい。ここでは核抽出液のみを調製する場合の一連の流れと、通常の方法と当研究室の方法で異なる点について述べる。以下の操作は氷上もしくは4℃で行っている。
核抽出液の調製
15 mLチューブに回収した細胞を液体窒素で凍らせる(−80℃でストック可)。低張液(当研究室ではKClの濃度を0.5 mMにしている)で解凍した細胞を再懸濁し細胞を膨張させる。ダウンス型ホモジェナイザー(Wheaton,B-pestle)で10〜20回ストロークし、細胞を破砕する。
細胞抽出液を15 mLチューブに移し、2,000 rpmで10分間遠心する。上清を捨て、ペレットを新しいバッファーで再懸濁した後、超遠心チューブに移し、MLS50ローター(ベックマン・コールター)で18,000 rpm(25,000 gav)、20分間遠心する。上清を除き、沈殿した核画分に0.42 M NaClを含むバッファーを加え、再び10回程度ストロークし、核画分をホモジェナイズする。
粗核抽出液を15 mLチューブに移し、30分間、ローテーションしながらインキュベーションする。インキュベーション後、超遠心チューブに移し、18,000 rpm(25,000 gav)で30分間遠心する。遠心後の上清をその上清の50倍以上の透析バッファー(当研究室ではIPバッファーを透析バッファーとして用いている)で数時間(2時間以上)透析する。新しい透析バッファーに置換しさらに数時間透析を行う。透析後のサンプルを1.5 mLチューブに移し、15,000 rpmで20分間遠心する。上清を新しいチューブに移し、タンパク質濃度を測定する。液体窒素で凍らせて、−80℃で保存する。
3. アフィニティー精製
以下実際のアフィニティー精製の手法について示すが、本プロトコールは当研究室で研究対象にしているMRG15と呼ばれる核タンパク質に最適化した方法であり、目的とするタンパク質に応じて精製条件を改変する必要があると思われる。また、今回示す3xFLAGタグとSBPタグによる精製は、使用する試薬や反応時間は異なるものの概ねの流れは同じである。また、精製の操作はすべて氷上もしくは4℃で行っている。
核抽出液(核タンパク質)約2 mg(コンフルエントになった15 cmディッシュ、約15枚から調製した核タンパク質量に相当)を使用した場合のプロトコールを以下に示す。
3-1. 3xFLAGタグ
3-1-1. プレクリア(前処理)4
200 µL(50%スラリー)のSepharose 4B5を1.5 mLチューブに入れ、IPバッファーで平衡化する。プロテアーゼインヒビター6を加えたIPバッファーで、核抽出液を2 mg/mLに希釈する。遠心(スイングローターの場合は1,500 rpmで3分間、アングルローターの場合は4,000 rpmで30秒間遠心後、チューブの背側の向きを逆にしてさらに30秒間遠心する7(以下レジンを遠心する場合同様の条件で行うこととする)。沈殿させたSepharose 4Bに希釈したサンプル1 mLを加え、ローテーションしながら30分間インキュベーションする。インキュベーションの後、遠心し、レジンを沈降させ上清を次のステップに用いる8。
3-1-2. アフィニティーレジンとの結合
50 µL(50%スラリー)のアフィニティーレジン(ANTI-FLAG M2 Affinity Gel)をIPバッファーで平衡化した後、遠心し、上清の平衡化したバッファーを取り除く。アフィニティーレジンの入ったチューブにプレクリアした後の上清をSepharose 4Bを吸い取らないように移し、3時間以上、ローテーションしながらインキュベーションする9。
3-1-3. レジンの洗浄
インキュベーション後のアフィニティーレジンを遠心し、上清を取り除いた後、新たに1 mLのIPバッファーを添加し、軽く転倒混和してレジンを懸濁してから遠心する。再び、上清を取り除き、新しいIPバッファーを加え、遠心する。この一連の洗浄操作を少なくとも3回以上繰り返す10。
3-1-4. 溶出
遠心し上清を取り除いた後、溶出バッファー(250 µg/mL 3xFLAG peptide)200 µLを加え、2〜3時間ローテーションすることで結合したタンパク質を溶出する11。遠心してレジンを沈降させ、上清を精製産物として回収する。レジンに残って溶出されないままのタンパク質をチェックするため、溶出バッファーと等量のSDSサンプルバッファーを加えレジンをボイルし、レジンフラクション(Resin)として回収する。
3-2. SBPタグ
3-2-1. プレクリア(前処理)4
200 µL(50%スラリー)のSepharose 4B5を1.5 mLチューブに入れ、IPバッファーで平衡化する。プロテアーゼインヒビターを加えたIPバッファーで、核抽出液を2 mg/mLに希釈する。遠心し、沈殿させたSepharose 4Bに希釈したサンプル1 mLを加え、ローテーションしながら30分間インキュベーションする。インキュベーションの後、遠心し、レジンを沈降させ上清を次のステップに用いる8。
3-2-2. アフィニティーレジンとの結合
50 µL(50%スラリー)のアフィニティーレジン(Streptavidin Sepharose)をIPバッファーで平衡化した後、遠心し上清の平衡化したバッファーを除く。アフィニティーレジンの入ったチューブにプレクリアした後の上清をSepharose 4Bを吸い取らないように移し、1〜3時間、ローテーションしながらインキュベーションする9。
3-2-3. レジンの洗浄
インキュベーション後のアフィニティーレジンを遠心し、上清を取り除いた後、新たに1 mLのIPバッファーを添加し、軽く転倒混和してレジンを懸濁してから遠心する。再び、上清を取り除き、新しいIPバッファーを加え、遠心する。この一連の洗浄の操作を少なくとも3回以上繰り返す10。
3-2-4. 溶出
遠心し、上清を取り除いた後、溶出バッファー(2 mM Biotin)200 µLを加え、30分〜1時間ローテーションすることで結合したタンパク質を溶出する11。遠心してレジンを沈降させ、上清を精製産物として回収する。レジンに残って溶出されないままのタンパク質をチェックするため、溶出バッファーと等量のSDSサンプルバッファーを加えレジンをボイルし、レジンフラクション(Resin)として回収する。
4. 精製産物の確認
まずウェスタンブロットによってそれぞれのタグを用いた場合の精製効率の比較をした(図4)。3xFLAGタグによる精製ではフロースルー(図4A、レーン4)に残ったものがほとんどないことから、タグとレジンとの親和性が非常に高いことがわかる。しかし、そのため溶出後のレジンに残ったままのものが多くなっている(図4A、レーン8)。また、SBPタグはレジンとの親和性は3xFLAGほどではないものの(図4B、レーン4)、高い溶出効率を得る事ができた(図4B、レーン6、8)。次にSDS-PAGE後のサンプルの銀染色による比較を行った(図5)。どちらも相互作用因子を特異的に精製できていることがわかったが、シングルの精製ではSBPタグの方が非特異的に結合したタンパク質が少なく、精製度は高いように思われる(図5B、Single)。
3xFLAG-SBP-MRG15を含む複合体をStreptavidin Sepharoseで精製した後、その溶出画分を再度anti-FLAG M2 affinity gelで精製したところ、特異的に結合するタンパク質は確認できるものの、相互作用する因子は減少する事が分かった(図5B、Tandem)。本研究では核抽出液をスタートの材料に用いたため、Singleの精製のみで十分な精製が可能になったのではないかと考えられる。またanti-FLAG affinity gel由来と考えられる非特異結合のタンパク質が強く検出された。これは一段階目の溶出タンパク質量に比べて用いたaffinity gelの容量が多かったのが原因と考えられる。また結合するタンパク質の種類が減少したのは、3xFLAG-SBP-MRG15と弱く結合するタンパク質が、2段階目の洗浄操作で遊離したためと考えられる。SBPとFLAGの順序を変えた場合、さらに結果が改善される可能性も考えられる。
それぞれのタグの特徴を踏まえて、現在当研究室では3xFLAGタグはよりスケールを上げて精製を行う場合に、SBPタグは簡便に精製度の高いサンプルを得るような場合に、両者を組み合わせた2段階精製は1段階だけではバックが高いような場合、というような使い分けを考えている。また、3xFLAG-MRG15の細胞株で、さらにスケールを上げた精製を行い、質量分析で相互作用因子を同定したところ、以前に同定したFLAG-MRG15のものと比べ、より少ない核抽出液からいくつかの新規の因子(MRG15を含む複合体の構成因子であることは報告されていた因子)を同定する事ができた。このことより3xFLAGタグは単独のFLAGタグより精製効率が高いことがわかった。
-
哺乳類用発現ベクターは目的の細胞株を単離するための選択マーカー(例:ゼオシン、ネオマイシン、ピューロマイシン、G418等)が組み込まれていれば何を使用してもよい。当研究室では誘導発現可能なT-REX system(Invitrogen)を用いており、Tetオペロンを有するpcDNA4/TOベクターにタグを融合したタグベクターを作製してから、目的の遺伝子を組み込んでいる。 ↩
-
本実験ではTetリプレッサーを安定に発現するHeLa細胞(T-REX HeLa)を用いた。 ↩
-
タンパク質の種類、発現量、解析の目的にもよるが、銀染レベルで複合体を確認するのであれば15 cmのシャーレ10枚以上の細胞を用いるのが望ましい。 ↩
-
レジンへ非特異的結合するタンパク質を除去するため行っている。細胞内に多量に存在する因子の精製などの場合は省略も可。アフィニティーレジンと同じ種類のレジンを使う方が望ましい。 ↩ ↩2
-
レジンの結合キャパシティーはそれぞれ0.6 mg/mL(FLAG-BAP),6 mg/mL(ビオチン化血清アルブミン)と記載されているが、本実験で用いている3xFLAGやSBPタグでは結合キャパシティーは異なると思われる。標的蛋白質の種類や大きさ、発現量によっても変化するため、予備実験によってレジンの最適容量を評価するのが望ましい。 ↩ ↩2
-
PMSFや市販のprotease inhibitor(Complete,Rocheなど)を混合して使用する。 ↩
-
2段階の遠心操作は遠心後のレジンを安定化させるために行っている。 ↩
-
最初の上清は結合しなかったフラクションとして回収する。また、洗浄は回数を重ねるほど精製度が上がるが、逆にロスが多くなる場合がある。 ↩ ↩2
-
銀染色で検出する時など、濃縮してから解析するような場合、最初の溶出後のレジンに新たな溶出バッファーを添加し、複数回溶出操作を繰り返す方が良い。 ↩ ↩2
工夫とコツ
精製条件
タンパク質の種類によって最適な条件は異なるため、その条件検討が免疫沈降(アフィニティー精製)の成功の鍵になると考えられる。どの条件を変えるかは経験的な要素が必要になるかもしれないが、簡単に変更できる条件としてバッファーに使用する塩の濃度、種類(例えばNaClかKCl)、界面活性剤の有無、濃度、種類、各反応の時間などがある。また、精製効率を高める上で重要なポイントは洗浄操作である。途中でレジンを吸ってしまうとロスが多く、逆に十分に洗浄できていないとバックが高くなる。カラム(Poly-Prep Column,BIO-RADなど)にレジンを移して洗浄する方がより精製度を上げることができるが、ある程度のレジンの量がないと逆にロスしてしまうのと、洗浄に時間がかかってしまうといった欠点がある。
アフィニティータグ
精製条件を色々検討しても精製効率が改善しない場合、タグとタンパク質の相性が悪いことが考えられる。その場合、タグの位置(N末端、C末端)や種類を変えてみるのも一つの手段である。
トラブルシューティング
本稿で記載したタンパク質の精製方法では、実際に相互作用しているタンパク質の有無をデータとして確認できるまで数多くのステップが存在することから、目的としている標的タンパク質が確実にアフィニティーレジンに結合しているのか、またレジンから溶出されているか、逐一サンプルを取り分けておき、ウェスタンブロット等によってそれぞれのステップでの状態を確認しながら進めることが大事である。アフィニティータグの特性は実験条件によって大きく変化することは無いと思われるので、精製の効率が上がらないような場合、あるいはバックグランドが高いような場合は、反応させる時間、洗浄の条件などを変化させて改善する必要があると思われる。
文献
- Terpe, K., et al., Appl. Microbiol. Biotechnol., 60, 523–533 (2003)
- Rigaut, G., et al., Nat. Biotechnol., 17, 1030–1032 (1999)
- Wilson, D. S., et al., Proc. Natl. Acad. Sci. USA, 98, 3750–3755 (2001)
- Hayakawa, T., et al., Genes Cells., 12, 811–826 (2007)
- Nishibuchi, G., et al., J. Biol. Chem., 289, 28956–28970 (2014)
- Dignam, J., et al., Nucleic Acids Res., 11, 1475–1489 (1983)
-
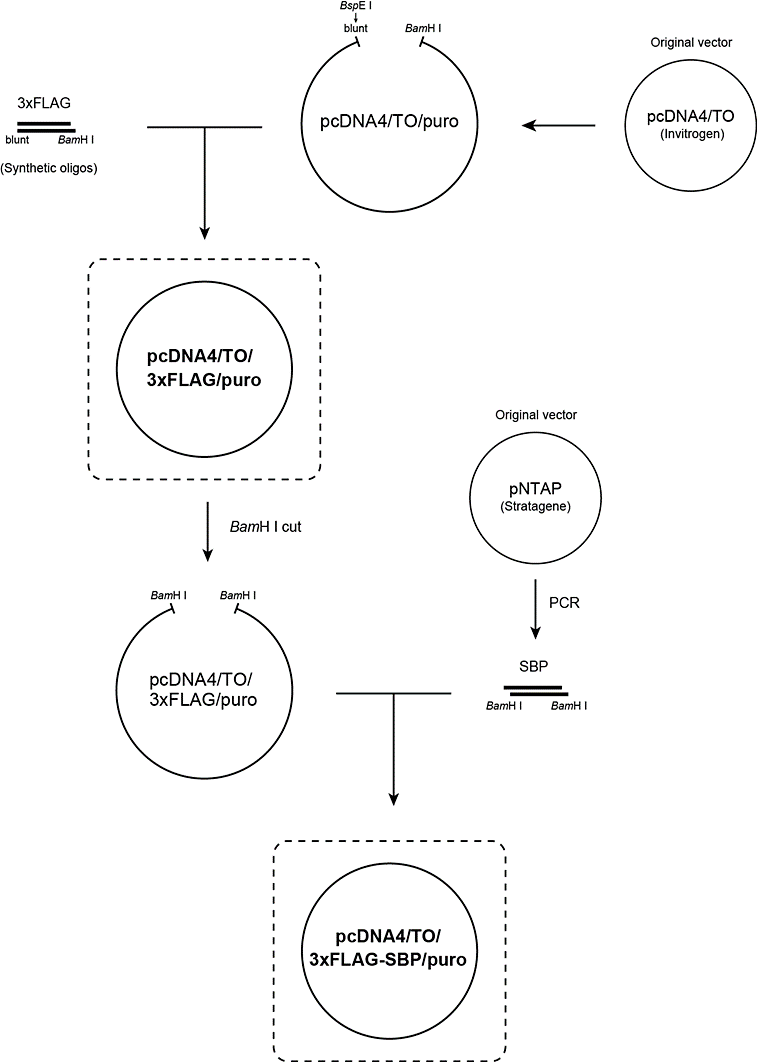
図1:タグを融合するための発現ベクターの構築模式図 -
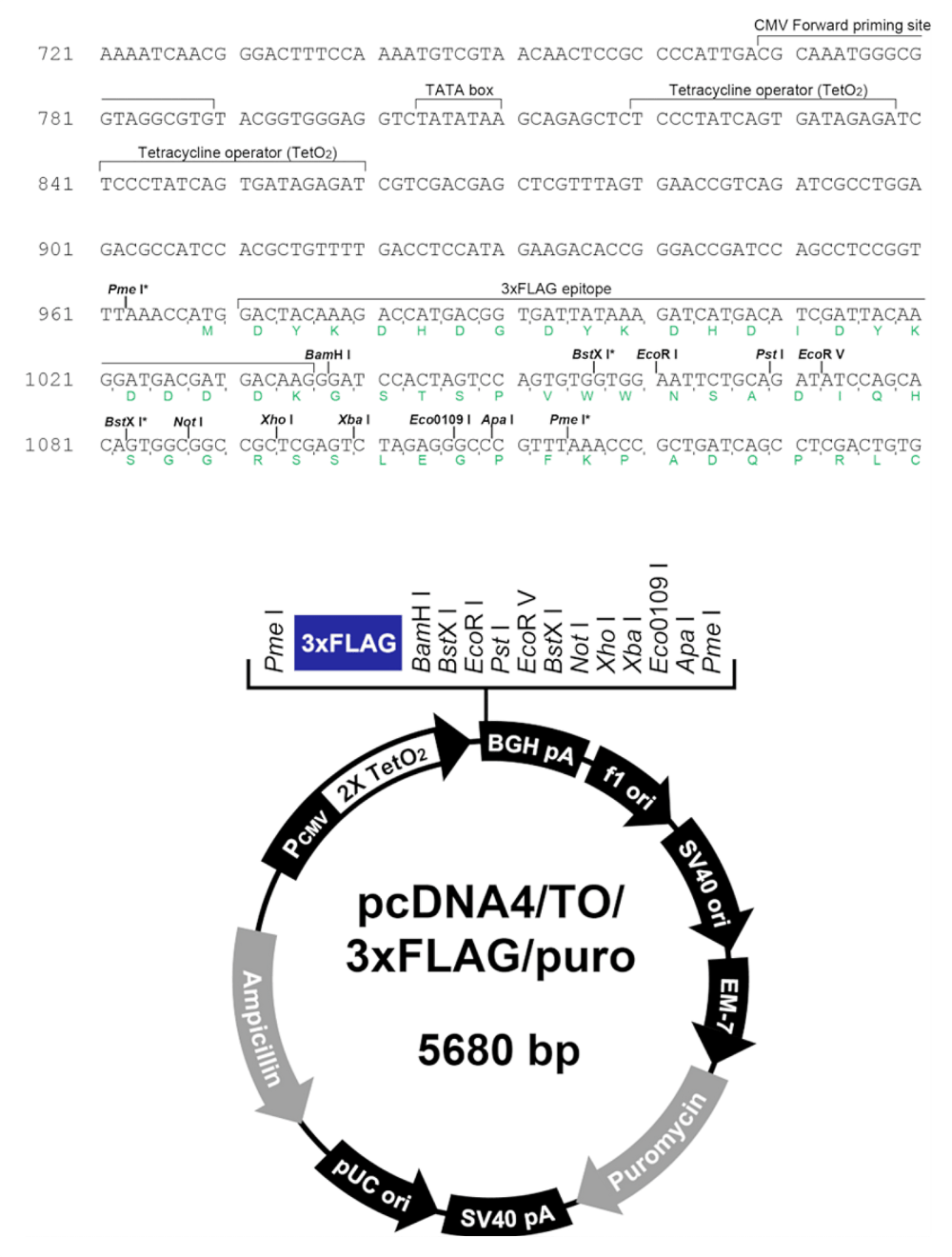
図2:本研究で用いたpcDNA4/TO/3xFLAG/puroの模式図 -
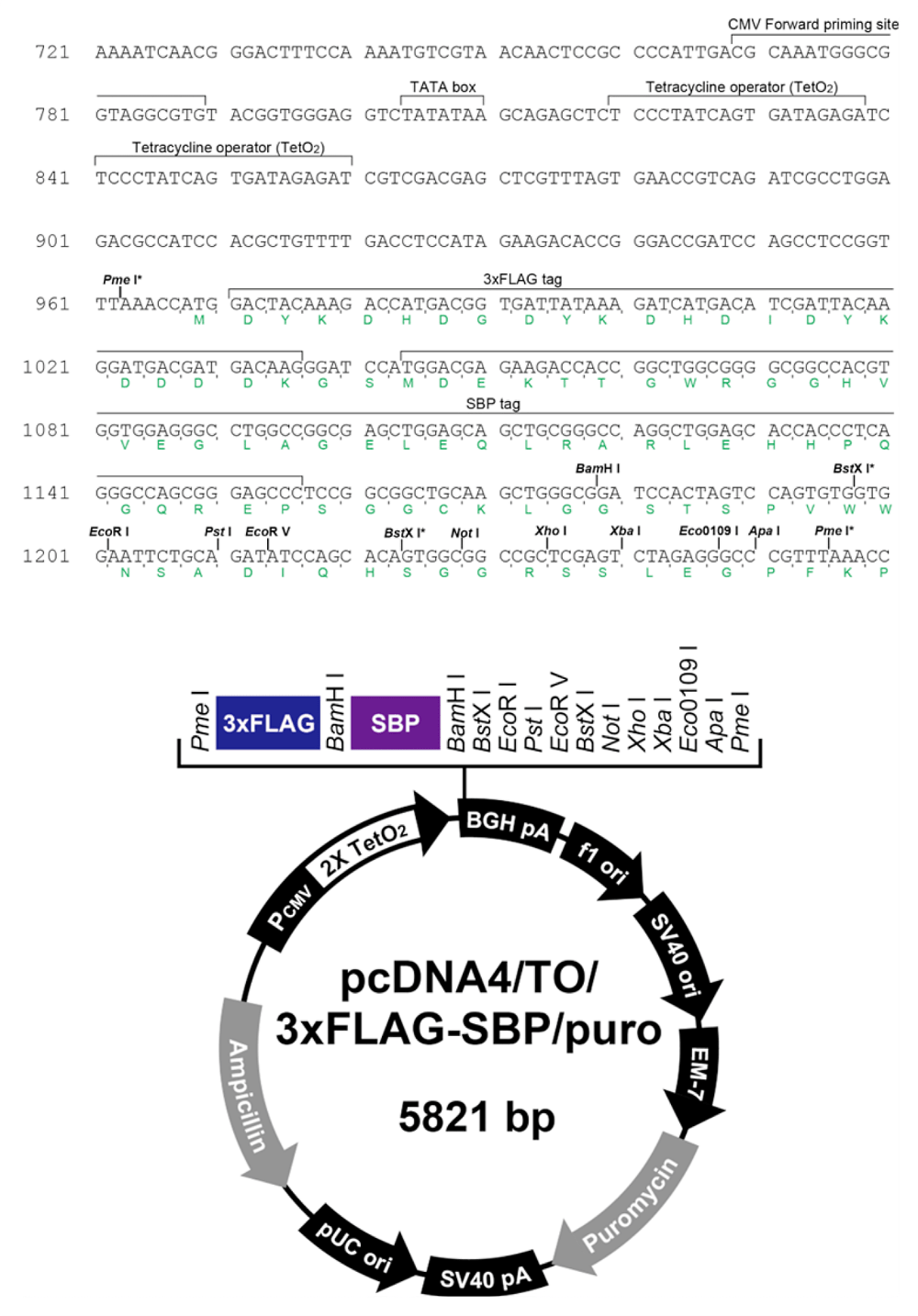
図3:本研究で用いたpcDNA4/TO/3xFLAG-SBP/puroの模式図 -
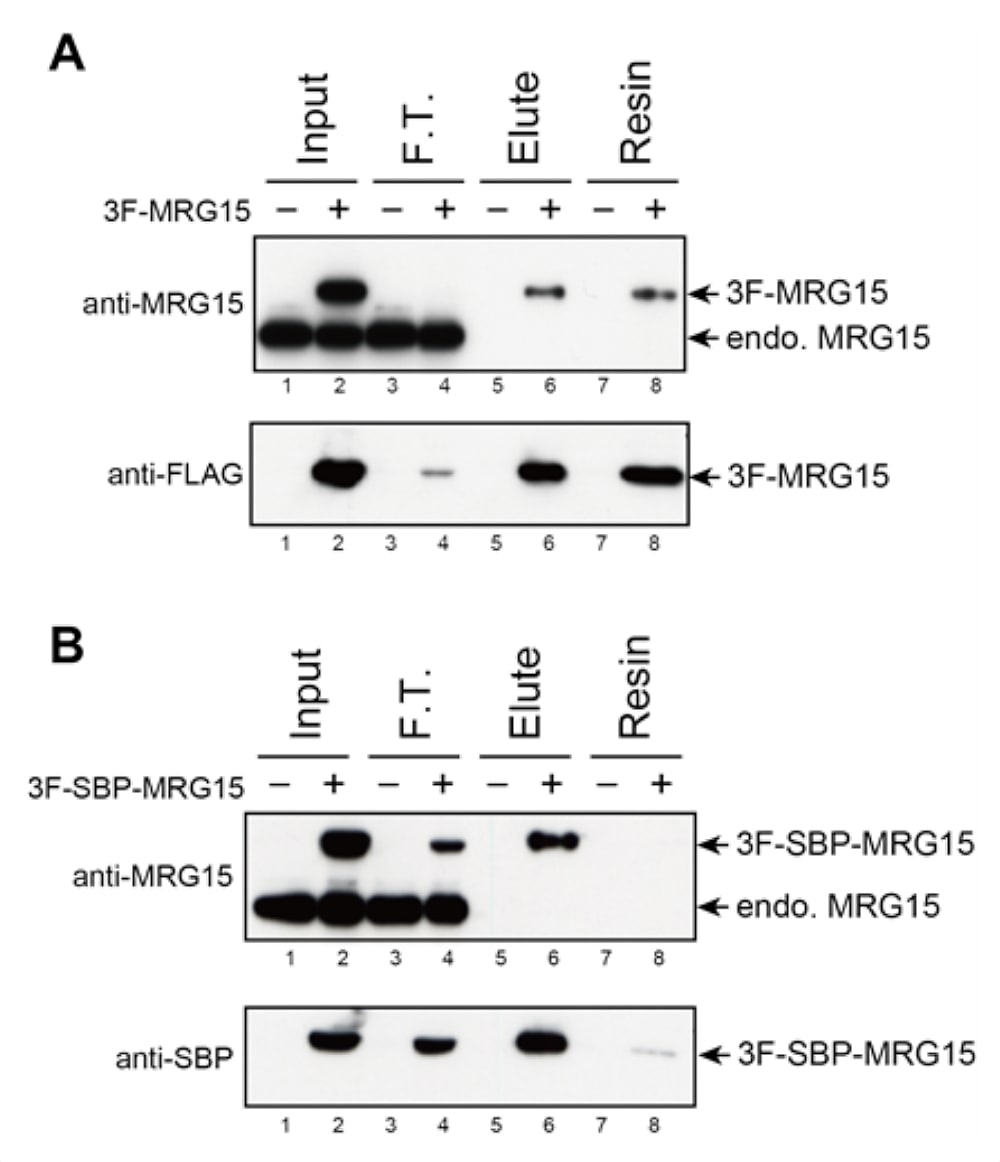
図4:各精製段階の結合溶出状態を確認したウェスタンブロット
(A)3xFLAGタグを付加したMRG15をanti-FLAG M2 affinity gelで精製。(B)3xFLAG-SBPタグを付加したMRG15をStreptavidin Sepharoseで精製。コントロールベクターを安定導入させた細胞株をネガティブコントロールとして用いた。インプット(Input)、非結合画分(Flow through,F.T.)、溶出画分(Elute)、レジンへの残留画分(Resin)。 -
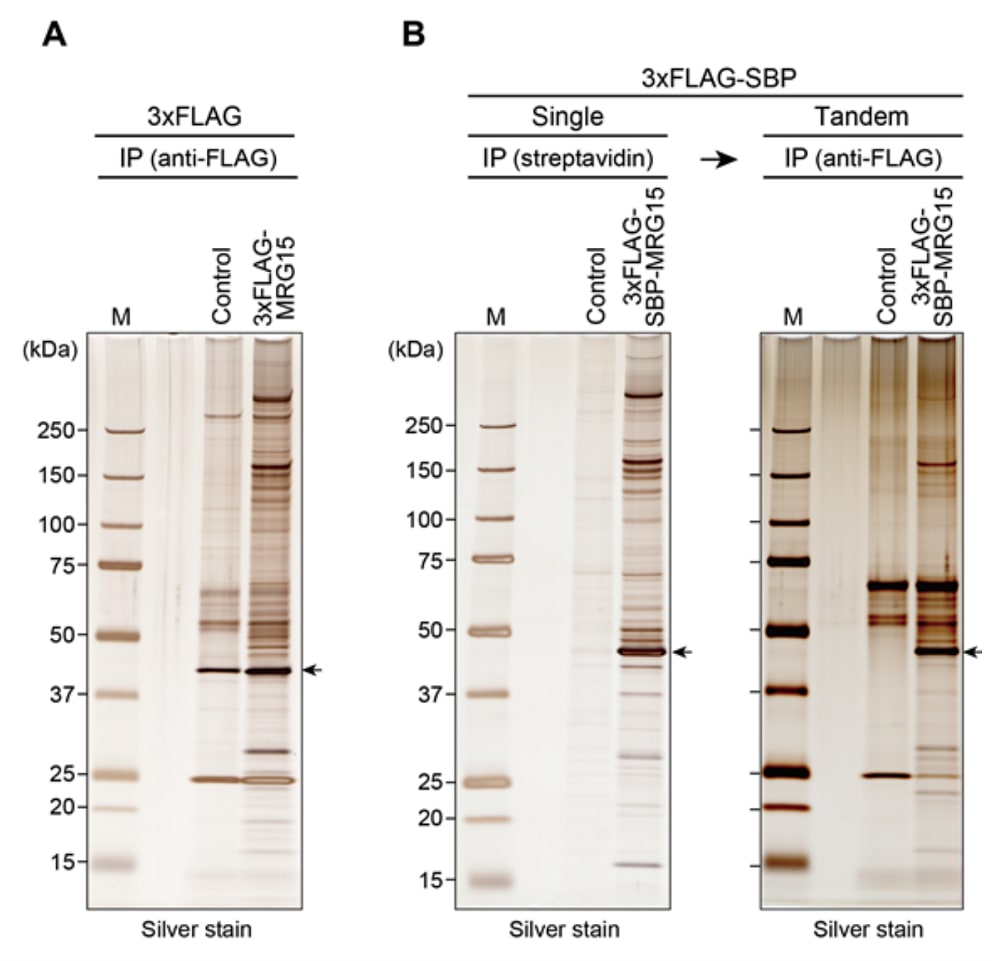
図5:精製されたタンパク質の確認
(A)3xFLAGタグを付加したMRG15をanti-FLAG M2 affinity gelで精製。(B)3xFLAG-SBPタグを付加したMRG15をStreptavidin Sepharoseで精製(左、Single)。さらに溶出後のサンプルを再度anti-FLAG M2 affinity gelで精製(右、Tandem)。コントロールベクターを安定導入させた細胞株をネガティブコントロールとして用いた(Control)。矢印はそれぞれ溶出された3xFLAG-MRG15, 3xFLAG-SBP-MRG15を示している。
