

概要
2024年のノーベル化学賞に象徴されるように、Google DeepMind社が開発したAlphaFold2(AF2)はタンパク質構造予測に革命を起こした。本稿を執筆している2024年11月には最新版であるAlphaFold3(AF3)もオープンソース化され、リガンドや核酸との複合体構造も非商業目的であれば自由に予測を行えるようになった。AF2やAF3の解説や基本的な利用方法は多くのWebページや書籍で既にまとめられているので、本稿ではAF2を用いた複数の準安定状態をもつタンパク質の構造予測に焦点を当て、その手順をまとめる。AF2(おそらくAF3も)は予測にバイアスが存在することが知られており、複数状態を探索するには工夫が必要となる。ここではAF2での構造予測に用いられる多重配列アラインメントのサブサンプリング、また共進化情報をもとにした変異導入によって構造予測のバイアスを克服する方法を示す。
イントロダクション
AF2の登場以降、タンパク質の構造予測そしてデザインにおいてAIは中心的な役割を担うようになった。それを象徴するように、2024年のノーベル化学賞はタンパク質デザインで先駆的研究を行うDavid Baker氏(ワシントン大学)とともに、AF2を開発したGoogle DeepMind社のDemis Hassabis氏、John Jumper氏に授与されることとなった。AF2はアミノ酸配列から多重配列アラインメント(MSA)を構築し、このMSAに含まれる情報からニューラルネットワークを用いてタンパク質構造を高精度で予測する (1)。本稿を執筆している2024年11月には最新版であるAF3もオープンソース化され、リガンドや核酸との複合体構造も非商業目的であれば自由に予測を行えるようになった (2)。AF2やAF3によってタンパク質の構造的理解は今後ますます進んでいくだろう。
AF2は高精度のタンパク質構造予測を可能にし、構造生物学、構造バイオインフォマティクスに革命を起こしたが、弱点も指摘されていた。それが構造予測のバイアスである。タンパク質は多くの場合、複数の準安定状態を有し、それらの状態間を切り替えることで機能する。しかしAF2の予測は特定の構造状態に偏ってしまい、構造状態探索が困難とされてきた。このバイアスは学習データやクエリ配列から構築されるMSAにある偏りに起因すると考えられ、テンプレート構造の利用やMSAを改変する等によって克服する方法が提案されている (3)。
本稿ではトランスポータータンパク質を対象に著者らが取り組んできた内容をもとに、複数構造状態を効率的に探索するプロトコールをまとめる (4,5)。AF2やAF3の基本的な使い方は多くのWebページや書籍にまとめられているので(例えば (6))、複数構造状態をどのようにAF2で探索するかに重点を置き紹介する。なお、本稿ではAF2のみを取り扱うが、構造予測のバイアスはAF3にも依然として潜んでいることが報告されている (2)。そのためここで紹介する方法はAF3にも応用できる可能性はあるが、現時点では未検証である。現在もしくは今後AF3を利用される本稿の読者には、是非ここで学んだ方法やアイデアがAF3にも適用できるかどうか研究を進めていってほしい。
装置・ソフトウェア
AF2はGitHubからダウンロードしインストールすれば使用できるが、3テラバイト程度のディスク容量に加えGPUも必要となってくるので、自前の計算機で気軽に始めるにはハードルが高い。これを回避するには、例えばスパコンの利用が1つの手である。例えば、分子科学研究所の計算科学研究センターはAF2、AF3ともに導入済みのスパコンを保有しており、利用申請と審査を経ることで利用可能である。より気軽に始めるにはGoogle Colab版のAF2や、AF2のMSA構築を高速化したColabFold(CF)(7) を利用すれば良い。これらはクラウド環境で構造予測を行うので、ローカル環境にインストールする必要がなくすぐに予測を開始できる。CFでは後述するMSAサブサンプリングのためのパラメータを調整できる。また、CFをローカル環境で実行するLocalColabFoldは、本家のAF2と異なり大規模サイズのデータベースを用意する必要がなく、またGoogle Colab版のような利用時間等の制限がないため、大量の構造予測を行いたい場合などに便利である。
本稿で紹介するプロトコールではLocalColabFoldを利用した(以後、特に断りがない限りLocalColabFoldを指して略称CFを用いる)。導入手順はCFのGitHubページを参照されたい。CFはNVIDIA製のGPUを搭載したマシンであれば、Linux、Windows(WSLを利用)で高速に実施できる。また、複数構造状態を探索するための変異導入では、変異を導入するアミノ酸残基を決定するため、MSAのDirect Coupling Analysis(DCA)を行う。そのために必要なpythonライブラリとして、pydca (8) を導入しておく。
計算手順
- アミノ酸配列の用意
- ColabFoldによる構造予測
- MSAサブサンプリング
- Direct Coupling Analysisと変異導入
計算の詳細
1. アミノ酸配列の用意
構造を予測したいタンパク質のアミノ酸配列がAF2/CFの入力となる。アミノ酸配列はUniProtデータベース等から入手できる。ダウンロードされたファイルはfasta形式となっており、これをそのまま次のCFの入力に利用できる。なお多くのタンパク質では、UniProtのStructure欄にX線結晶構造解析等で得られた既知構造とともにAF2によって予測された構造も記載されている。こうしたAF2予測構造はAlphaFold Structure Database (9) にまとめられている。構造予測を始める前にまずこの情報も見てみると良いだろう。
2. ColabFoldによる構造予測
CFは以下のようにして実行できる。
$ colabfold_batch --num-seeds 20 --max-msa 512:1024 input.fasta ./output_dir/
input.fasta が上記1で入手したfasta形式のアミノ酸配列であり、予測結果は output_dir 下に保存される。output_dir に保存される出力は構造ファイル(PDB形式)と予測スコアである(予測スコアについては次の3で言及する)。ここで用いたオプションの意味は以下の通りである(ここでは用いていないオプションについてはCFのGitHubページを参照のこと)。
--num-seed:乱数の種の数を指定する。上の例では20を指定。乱数の種ごとに5つの予測構造モデルが出力されるので、ここでは合計100構造が得られる。--max-msa:MSAの中から実際に構造予測に使用されるアミノ酸配列の数(これを “MSAの深さ” と表現することがある)を指定する。2つの整数を指定し、前者はMSAをクラスタリングした後に実際に予測に用いられるクラスターの数、後者はそれとは別に追加で選ばれ構造予測時の情報として用いられる配列の数である。小さな値を設定すると “浅い” MSAを用い構造予測が行われる。
例として、硝酸と亜硝酸を輸送するトランスポーターNarKのアミノ酸配列(UniProt ID: P10903、アミノ酸残基数:463)に対し構造予測を行う場合、著者の計算環境(GPU: NVIDIA A4000, CPU: Intel Core i7-12700)では2時間弱で100構造が得られる。
3. MSAサブサンプリング
AF2/CFではクエリ配列からMSAを構築し、MSAに含まれる類縁タンパク質のアミノ酸配列集団をランダムにサブサンプリングして構造予測を行う。MSAサブサンプリングによって集める配列数(MSAの深さ)は、CFのオプション --max-msa によって指定される。AF2/CFはMSAから得られるアミノ酸残基間の共進化情報を構造予測に利用しているので、MSAの深さを浅くすることで共進化情報の統計的不確かさが増大し、多様な構造状態が予測できるようになる (10)。
先に例として挙げたNarKについて、オプション--max-msa の値を 512:1024、256:512、128:256、64:128、32:64、16:32、8:16 と減らしていったときの予測構造の分布を図1に示す (5)。トランスポータータンパク質は外開き、閉、内開きの三構造状態を交互に切り替えることで基質を輸送するが、深いMSA(例えば 512:1024)では内開き構造のみが予測されてしまう。一方で、MSAを浅くしていくと閉構造や外開き構造も含んだ幅広い構造状態が予測されていることがわかる。なお、この例のNarKでは外開き構造は実験的に得られておらず、CFによって未解明構造も予測できることを示している。予測構造の信頼性は、CFが出力するいくつかのスコアによって評価できる。例えばpLDDTスコアはアミノ酸残基周囲の局所環境の信頼性を0から100までの数値で表し、値が大きいほど信頼性が高いことを示す。NarKについて予測された外開き構造を図2で示し、pLDDTスコアに従って各残基を色付けしている。N末端やC末端など柔軟性が高く特定の構造をとらないような領域を除いて、高い信頼度で予測構造が得られていることがわかる。
CFの構造予測にバイアスがある際、どの程度MSAを浅くすれば良いかは決まった指針があるわけでなく、タンパク質ごとに異なる。そのため手探りで幾通りかの深さを試す必要がある。このときMSAを毎回構築し直す必要はなく、始めに作ったMSAを次回以降は利用できる。CFではa3m形式のMSAファイルを出力するので、MSAの深さを変えて次に予測を行う際は、先述の2で示した colabfold_batch コマンドにおいて input.fasta をこのa3m形式のMSAに置き換えれば良い。
一般にはより浅いMSAを使うほど予測構造の多様性が増加する。しかし浅いMSAでは統計的不確かが増大するため、信頼性の低い構造が予測される可能性があることに注意が必要である。NarKの構造予測を例にすると、試みた中で最も浅いMSA(--max-msa 8:16)では膜貫通ヘリックスが部分的にほどけた構造(図3上段)や、誤ったトポロジーをもつ構造(図3下段)が出てきてしまう。そのためスコアと構造の確認を十分に行う必要がある。
4. Direct Coupling Analysisと変異導入
上記3では、MSAのサブサンプリングに含まれる統計的不確かによって共進化情報を乱し、複数構造状態を予測できるようにした。しかし、もしMSA内の共進化情報を事前に知ることができれば、高い共進化傾向によってバイアスをもたらしているアミノ酸残基ペアに変異を導入することで異なる構造状態を予測きるはずである。この方法を最後に紹介したい。
MSA内の共進化情報を抽出する方法はいくつかあるが、ここではDirect Coupling Analysis(DCA)を用いる。DCAは、pythonであればpydcaを導入することで簡単に実施できる (8)。pydcaはpython 3.5以降の環境であれば用いることができ、コマンドライン上で
$ pip install pydca
を実行することで導入できる(著者はpython 3.5.4、pydca 1.23を利用)。
CFのMSAはa3m形式で出力されるが、pydcaではFASTA形式を入力とする必要がある。クエリ配列には存在しない領域(挿入領域)について、a3m形式では類縁タンパク質のアミノ酸配列を小文字で表し、FASTA形式ではクエリ配列側に “-” を入れる(FASTA形式の場合、大文字と小文字の区別はない)。今回興味があるのはクエリ配列に存在する領域の中での共進化情報なので、挿入領域を考慮する必要がない。そのため例えばLinuxであれば
$ tr -d a-z msa.a3m | grep -v "#" > msa.fasta
などとすれば良い(a~zの小文字を削除し、#で始まるコメント行を削除してmsa.fastaに保存している)。
pydcaはpythonスクリプト内およびコマンドライン上で使用できるが、ここではコマンドラインで実行する方法を示す。
$ plmdca compute_fn PROTEIN msa.fasta --max_iterations 500 --num_threads 6 --apc
plmdcaは疑尤度最大化DCAを実施することを意味する(平均場DCAを用いるmfdcaもあるが、一般に疑尤度最大化の方が信頼性が高いとされる)。compute_fn は、残基間の共進化スコア計算にFrobeniusノルムを用いることを指定し、PROTEIN は入力となるMSA(msa.fasta)がタンパク質のアミノ酸配列の集合であることを指定している(pydcaはRNAの塩基配列も対象にできる)。オプション --max_iterations は疑尤度最大化のイテレーション回数の最大値、--num_threads はスレッド数、--apc は共進化スコアをaverage-product correlationによって補正することを意味する。それぞれの詳細については、文献 (8,11) を参照のこと。
pydcaの出力ファイルには1、2列目にペアとなる2つの残基番号、3列目に共進化スコアが記載され(図4)、正に大きなスコアをもつ残基ペアほど共進化傾向が強い。あとはこの結果をもとに共進化傾向の強い残基ペアに変異を導入すれば良いのだが、ここで注意すべきことがある。それは、図4に示すように高い共進化スコアをもつ残基ペアの多くは同じ二次構造内や同じドメイン内に存在することである。しかし例えばトランスポータータンパク質の場合には、外開き、閉、内開きの3構造状態でドメイン間の残基–残基コンタクトに顕著な変化が生じる。よって、共進化スコアが高く、かつ野生型配列で予測された構造状態に特有のコンタクト残基ペアに変異を入れなければ意味がない。構造状態特有のコンタクトを特定することは容易ではないが、実験や分子シミュレーション等の情報があれば、それと共進化情報を組み合わせ変異導入先を決めると良いであろう。
例えば図5に示すように、シュウ酸トランスポーターOxlTではAF2の構造予測が外開き構造に偏る。そこで外開き構造(および閉構造)に特有のコンタクト残基をMD計算によって特定し、その中から共進化スコアが高い残基ペアG140-D280(図4緑下線)に変異を導入した。G140-D280は外開き構造のとき水素結合を形成し安定化しているため、D280を疎水性残基に変更(ここではロイシンを選択)し水素結合が形成できない変異型とした。すると、D280L変異型では期待通り内開き構造が得られることがわかった(アラニン等、他の疎水性残基への変異でも同様の結果が得られる)。この変異では外開き構造を不安定化することで内開き構造の予測を可能にしたが、それとは異なり内開き構造を安定化するような変異を導入することもできる。図5に示すように、内開き構造に特有のコンタクト残基ペアをシステインに変異することで、CFはジスルフィド結合を形成させようと内開き構造を予測するようになる。
複数構造状態の探索の簡便性でいえば3で述べた浅いMSAをまず試みた方が良いであろう。しかし変異による方法は、単なる構造探索のみならず構造状態間の安定性を変化させる変異候補の情報を提供できるという利点がある。AF3も登場したことにより、タンパク質構造予測は今後複合体等のより大規模で複雑な対象へと広がっていくだろう。ここで示したプロトコールがそうした研究の一助になれば幸いである。
文献
- Jumper, J., et al., Nature, 596, 583–589 (2021)
- Abramson, J., et al., Nature, 630, 493–500 (2024)
- Sala, D., et al., Curr. Opin. Struct. Biol., 81, 10264 (2023)
- Ohnuki, J., et al., J. Phys. Chem. Lett., 15, 725–732 (2024)
- Ohnuki, J. & Okazaki, K.-I., J. Phys. Chem. B, 128, 7530–7537 (2024)
- 富井健太郎 編, AlphaFold時代の構造バイオインフォマティクス実践ガイド(実験医学別冊 最強のステップUPシリーズ), 羊土社 (2024)
- Mirdita, M., et al., Nat. Methods, 19, 679–682 (2022)
- Zerihun, M. B., et al., Bioinformatics, 36, 2264–2265 (2020)
- Varadi, M., et al., Nucleic Acids Res., 50, D439–D444 (2022)
- del Alamo, D., et al., eLife, 11, e7575 (2022)
- Ekeberg, M., et al., Phys. Rev. E, 87, 012707 (2013)
-
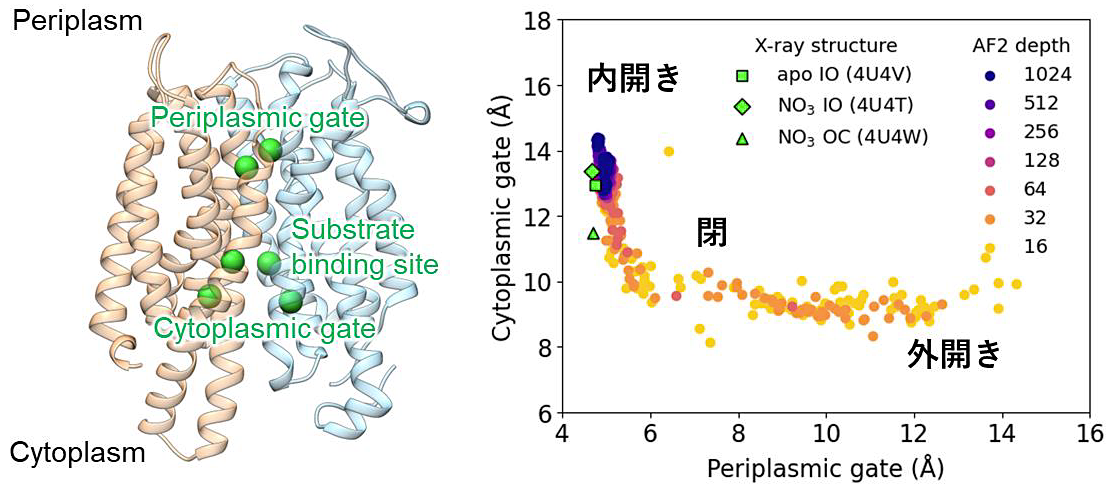
図1:CFによる硝酸・亜硝酸トランスポーターNarKの構造予測
NarKはperiplasm側、cytoplasm側それぞれに基質透過経路のゲートが存在し、periplasmゲートが開いた外開き構造、cytoplasmゲートが開いた内開き構造、両 ゲートが閉じた閉構造がある。MSAを浅くする構造予測に用いる(類縁タンパク質の配列数を減らす)ことで内開き構造のみならず、閉構造、外開き構造を含む多様な構造状態が予測できるようになる。 -
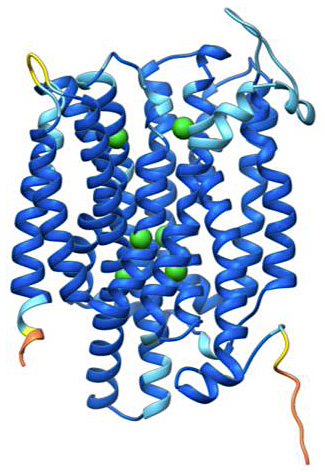
図2:NarKの外開き予測構造
pLDDTスコアに従ってアミノ酸残基を色付けしている(青:pLDDT ≥ 90;シアン:70 ≤ pLDDT < 90;黄:50 ≤ pLDDT < 70;橙:pLDDT < 50)。 -
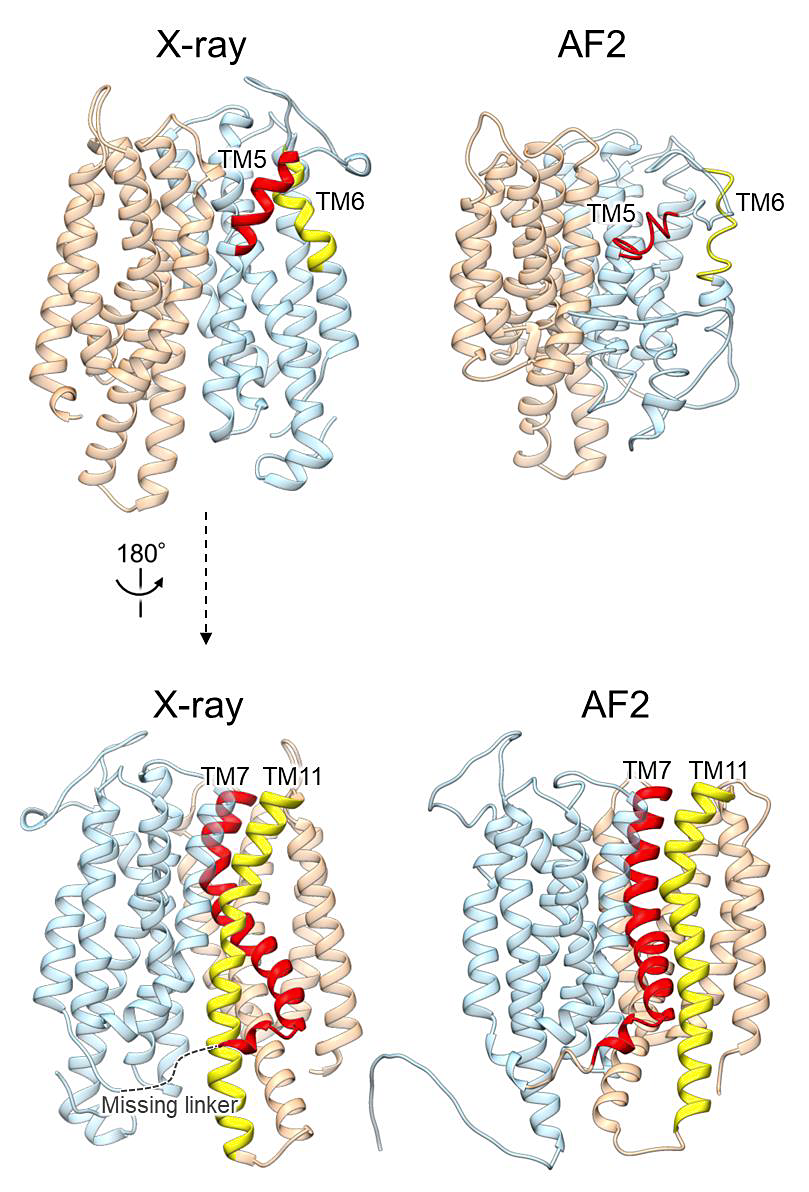
図3:最も浅いMSAから予測されたNarK予測構造
結晶構造と比べると、膜貫通ヘリックスが部分的にほどけている構造(上段)やトポロジーが正しくない構造(下段)が予測されてしまうことがある。 -
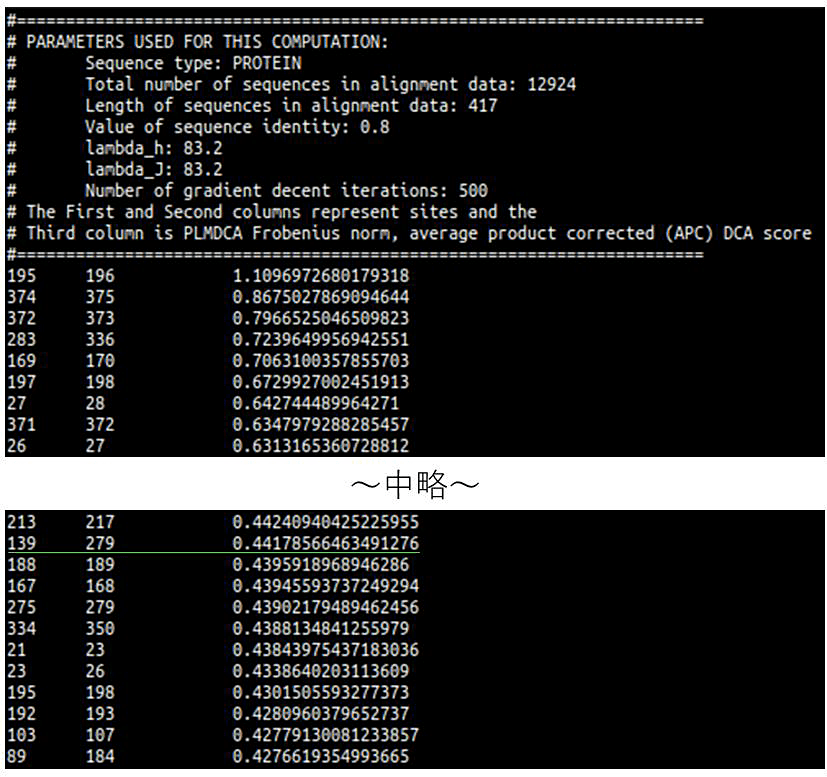
図4:pydcaの出力ファイル
1、2列目はペアとなる2つの残基番号、3列目が共進化スコアである。正に大きなスコアをもつアミノ酸残基ペアほど共進化傾向が強く、高スコアのものから順に記載されている。ここではOxlTのMSAを入力として得た結果の一部を示しており、翻訳開始メチオニンを除去したMSAを用いたため残基番号は1つずれている。高スコアをもつ残基ペアの大半は配列上で近く、タンパク質ドメイン内部の共進化傾向を示している。しかし一部には、緑下線で示すペア(G140–D280)のように配列上離れたドメイン間残基ペアも高スコアをもつ。 -
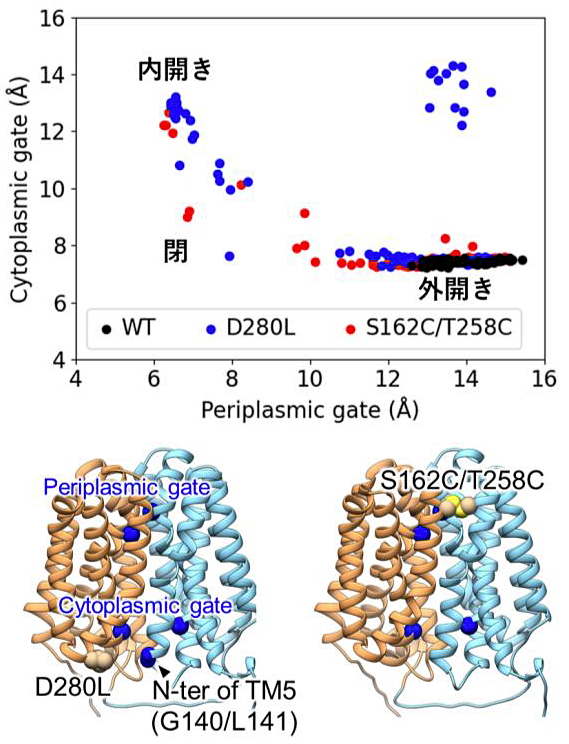
図5:CFによるシュウ酸トランスポーターOxlTの構造予測
外開き構造でドメイン間コンタクトを形成するD280(G140/L141とコンタクトを形成)は共進化スコアが高く、変異を導入することで内開き構造を予測するようになる。また、内開き構造でドメイン間コンタクトを形成するS162とT258をシステインに変異すれば、AF2はジスルフィド結合を予測し内開き構造が得られる。
