

概要
X 線結晶構造解析法によってタンパク質を構造解析するためには、まずそのタンパク質を結晶化する必要がある。そして結晶が得られれば、その結晶を用いて X 線回折実験を行い、X 線回折強度データを収集する。そこで得られた X 線回折強度データを用いて構造解析を行うが、この際に位相情報が必要となる。というのも、X 線回折強度データには構造解析に必要な位相情報が含まれないからである(詳細は、「X 線、位相問題」で検索し調べていただきたい)。その位相情報を得る手段として、分子置換法(MR 法)、重原子同型置換法(MIR 法)、多波長/単波長異常分散法(MAD/SAD 法)といった方法が用いられる。なかでも、類似のタンパク質の立体構造を使って位相情報を得る分子置換法はまず初めに試すべき方法であろう。なぜなら、2021年5月の時点で、Protein Data Bank Japan(https://pdbj.org/)には17万を超えるタンパク質の立体構造情報(PDB データ)が登録されているからで、今後構造解析されるタンパク質の多くは、類似のタンパク質の立体構造が登録されている可能性が高いからである。
さて、分子置換法は、1. 非対称単位中のタンパク質分子数の見積もり、2. 自己回転関数による非結晶学的回転軸の確認、3. 類似タンパク質(モデル分子)の選択と編集、4. モデル分子の回転/並進、5. 結晶中の分子のパッキングの確認という5段階に分けられる。これらは、CCP4 Program Suite (1) が導入されている Windows や Mac のパソコンさえあれば、1日で実施・完了できる。ただし、類似タンパク質とのアミノ酸同一性が低い場合(例えば20%以下の場合)には、4. の段階で容易に結果が得られずより多くの時間がかかることをご承知おき頂きたい。
ここでは初めて構造解析を実施する研究者を対象として、CCP4 Program Suite が導入されている Windows のパソコンを用いて、CCP4 Program Suite の Molrep (2) という分子置換法のソフトを用いた手順を解説する。
装置
CCP4 Program Suite(2021年5月現在 v7.1.013)と COOT が導入されている Windows パソコンを準備する。なお、Windows パソコンに CCP4 と COOT を導入するためには、「CCP4 Windows」で検索し、「CCP4 Program Suite including SHELX and COOT」をダウンロードした後、そのファイルをダブルクリックして「詳細」 を選択した後に実行するとよい。
実験手順
- 非対称単位中のタンパク質分子数の見積もり
- 自己回転関数による非結晶学的回転軸の確認
- 類似タンパク質(モデル分子)の選択と編集
- モデル分子の回転/並進
- 結晶中の分子のパッキングの確認
実験の詳細
本プロトコールを始める前に
目的タンパク質のアミノ酸配列が既知であり、X 線回折強度データファイル(MTZ File)が得られていることを前提として手順を解説する。
分子置換法の詳細
1. 非対称単位中のタンパク質分子数の見積もり
非対称単位とは、結晶中で行う対称操作により重ねることができない最小の領域のことを指す。なお、Protein Data Bank に登録されている立体構造情報は、通常は非対称単位に存在する全ての分子の情報を含んでいる。
非対称単位は、例えば空間群が P21 の場合には単位胞の 1/2 の大きさを持ち、空間群 が P43212 の場合には単位胞の 1/8 となる。このように空間群によって単位胞に対する非対称単位の割合が異なるので、詳しく知りたい場合には空間群の詳細が記載されている書籍(例えば、International Tables for Crystallography, Volume A, ISBN: 978-0-470-97423-0)をご参照いただきたい。
さて、分子置換法とは、類似タンパク質を回転・並進させ、非対称単位に存在する目的分子に対して類似タンパク質を重ねる操作法のことである。そのとき、非対称単位内に複数の分子が存在すれば、その分子全てに対して類似タンパク質を重ねる必要がある。したがって、まず非対称単位に何分子が含まれているかを以下の手順で見積もることから始める。
- CCP4i を起動し、Project 名と X 線回折強度データファイル(MTZ File)が存在するフォルダ設定した後に、CCP4i のメインパネルから Molecular Replacement モジュールを選択する(図1)。
- Analysis → Cell Content Analysis を選択すると Matthews - Cell Content Analysis パネルが起動する(図2)。
- Matthews ‒ Cell Content Analysis のパネルにて、MTZ file を Browse から選択すると、空間群、格子定数、分解能が自動で表示される。
- Molecular weight of protein or nucleic acid の欄に分子量を入力する。
- Run Now を選択する。
以下のような結果が表示される(以下は一例)。
Nmol/asym Matthews Coeff %solvent P(2.60) P(tot)
1 10.04 87.76 0.00 0.00
2 5.02 75.51 0.00 0.00
3 3.35 63.27 0.10 0.11
4 2.51 51.03 0.58 0.57
5 2.01 38.79 0.30 0.31
6 1.67 26.54 0.00 0.00
7 1.43 14.30 0.00 0.00
8 1.26 2.06 0.00 0.00
通常、表の Matthews Coeff で示されている値は 1.7~3.5 A3/Da 程度とされるため (3)、この場合は、非対称単位中におおよそ3~5分子が存在すると予想される。この段階では、分子の数を厳密に決定する必要はなく、ある程度の可能性を検討できれば、次の段階に進む。
2. 自己回転関数による非結晶学的回転軸の確認
非対称単位に複数の分子が存在することがわかれば、次に自己回転関数を計算し、非結晶学的回転軸(NCS)の存在を確認する。
- Molecular Replacement モジュール内の Molrep – auto MR を選択する。
- Molrep パネルにて、Do Self rotation function を選択し、MTZ file を Browse から選択する(図3)。
- 必要であれば、SRF options 内の Select Chi sections を選択して、NCS の候補の角度を入力する。例えば、2量体が5つ集まった10量体が存在している分子では、分子内に2回軸が5本と5回軸が存在する。初期値で180度は設定されているので、設定されていない5回軸(360度を5で割った数字72)を Select Chi sections に入力すると良い。
- Run→Run Now を選択する。
1. の①で指定したフォルダに .ps という拡張子のついた Postscript ファイルが出力される。このファイルを Adobe Distiller で PDF ファイルに変換した後に開くと(Adobe Illustrator ではそのまま閲覧可能)、極座標で示された図が表示される(図4)。
図4では、2回軸の存在を示す Chi = 180 のセクションに5つの高いピーク(上図の赤矢印)が等間隔で存在しており、Chi = 72 のセクションに1つの高いピーク(上図の青矢印)が存在していることから、2量体が5つ集まった10量体が存在している可能性が予想される。この自己回転関数の計算結果と、1. で見積もった非対称単位内の分子数とを比較して、結晶中で分子が何分子どのように配置しているかを予想しておく。
ただし、この 2. は分子内に NCS が存在して、なおかつそれが結晶学的回転軸とは異なる場合にのみ有用となる。そのため、図4のような明解な情報が得られない場合も多く、その場合は気にせず次の段階に進む。
3. 類似タンパク質(モデル分子)の選択と編集
まず、Protein Data Bank に登録されているタンパク質の中で、目的タンパク質とアミノ酸同一性の高いタンパク質(モデル分子)を選択する。モデル分子の選択は様々な方法があるが、以下では、Protein Data Bank から同一性の高いものを選択する方法を示す。
- https://www.rcsb.org/ にアクセスし Search→Sequence Search→Advanced Search - Sequence Search を選択する(図5)。
- Sequence? の空欄にアミノ酸配列(一文字記号)を入力する。
- Display Results as で Polymer Entities を選択し、虫眼鏡のマークを選択する。
- Sequence identity と E-value を確認する。私見であるが Sequence identity が20%以上あれば分子置換法を試す価値はあると思われる。Download Alignment を選択して Alignment ファイルをダウンロードしておく。その Alignment ファイルは、CLUSTALX というソフト等で表示できる。
- Sequence identity が高く E-value が小さい PDB ID を選択し、Download Files から PDB Format を選択して PDB ファイルをダウンロードし(図6)、そのファイルを 1. の①で指定したフォルダに保存する。
次に、テキストエディタ(例えば、Windows アクセサリのメモ帳)で PDB ファイルを開いてファイルの内容を確認する。PDB ファイルの主な行を説明すると、HEADER で始まる行には PDB ID が示され、CRYST1 には単位格子と空間群の情報が書かれている。ATOM にはタンパク質の座標データが記され、HETATM にはタンパク質に結合している基質アナログや水分子などの座標データが書かれている。
HEADER CELL CYCLE 05-NOV-16 5H5G
TITLE STAPHYLOCOCCUS AUREUS FTSZ-GDP IN T AND R STATES
COMPND MOL_ID: 1;
COMPND 2 MOLECULE: CELL DIVISION PROTEIN FTSZ;
(略)
CRYST1 43.917 159.023 44.058 90.00 92.78 90.00 P 1 21 1 4
ORIGX1 1.000000 0.000000 0.000000 0.00000
ORIGX2 0.000000 1.000000 0.000000 0.00000
ORIGX3 0.000000 0.000000 1.000000 0.00000
SCALE1 0.022770 0.000000 0.001104 0.00000
SCALE2 0.000000 0.006288 0.000000 0.00000
SCALE3 0.000000 0.000000 0.022724 0.00000
ATOM 1 N MET A 11 70.851 24.422 76.500 1.00 58.77 N
ATOM 2 CA MET A 11 69.861 23.493 75.871 1.00 60.16 C
ATOM 3 C MET A 11 70.402 23.013 74.526 1.00 54.98 C
ATOM 4 O MET A 11 70.878 23.819 73.724 1.00 52.56 O
ATOM 5 CB MET A 11 68.489 24.170 75.715 1.00 65.01 C
ATOM 6 CG MET A 11 67.383 23.242 75.219 1.00 70.22 C
ATOM 7 SD MET A 11 65.810 23.448 76.090 1.00 79.56 S
ATOM 8 CE MET A 11 64.628 22.796 74.912 1.00 76.98 C
ATOM 9 N ALA A 12 70.336 21.703 74.294 1.00 51.77 N
ATOM 10 CA ALA A 12 71.092 21.069 73.206 1.00 46.93 C
ATOM 11 C ALA A 12 70.521 21.394 71.823 1.00 42.06 C
ATOM 12 O ALA A 12 69.319 21.313 71.612 1.00 42.26 O
ATOM 13 CB ALA A 12 71.164 19.568 73.419 1.00 47.58 C
(略)
TER
ATOM 2229 N HIS B 10 30.445 -18.763 57.180 1.00 84.67 N
ATOM 2230 CA HIS B 10 29.963 -18.655 55.760 1.00 88.20 C
ATOM 2231 C HIS B 10 31.005 -19.137 54.736 1.00 83.06 C
ATOM 2232 O HIS B 10 30.737 -20.057 53.965 1.00 80.48 O
ATOM 2233 CB HIS B 10 29.535 -17.216 55.446 1.00 89.27 C
ATOM 2234 CG HIS B 10 29.024 -17.028 54.051 1.00 92.35 C
ATOM 2235 ND1 HIS B 10 27.735 -17.348 53.681 1.00 94.12 N
ATOM 2236 CD2 HIS B 10 29.628 -16.553 52.937 1.00 93.17 C
ATOM 2237 CE1 HIS B 10 27.565 -17.076 52.400 1.00 93.87 C
ATOM 2238 NE2 HIS B 10 28.699 -16.591 51.925 1.00 96.73 N
ATOM 2239 N MET B 11 32.176 -18.498 54.738 1.00 78.54 N
ATOM 2240 CA MET B 11 33.291 -18.831 53.841 1.00 75.35 C
ATOM 2241 C MET B 11 34.497 -19.169 54.713 1.00 66.55 C
ATOM 2242 O MET B 11 34.821 -18.415 55.632 1.00 67.02 O
ATOM 2243 CB MET B 11 33.608 -17.626 52.948 1.00 80.23 C
ATOM 2244 CG MET B 11 34.364 -17.949 51.667 1.00 84.89 C
ATOM 2245 SD MET B 11 33.314 -18.347 50.247 1.00 90.91 S
ATOM 2246 CE MET B 11 32.482 -16.780 49.969 1.00 92.25 C
(略)
TER 4475 PHE B 315
HETATM 4476 PB GDP A 401 73.156 10.830 45.052 1.00 29.42 P
HETATM 4477 O1B GDP A 401 74.218 10.572 44.048 1.00 33.95 O
HETATM 4478 O2B GDP A 401 72.385 12.106 44.954 1.00 30.71 O
HETATM 4479 O3B GDP A 401 73.676 10.647 46.448 1.00 36.84 O
HETATM 4480 O3A GDP A 401 72.122 9.629 44.803 1.00 30.55 O
G(略)
MASTER 319 0 3 26 23 0 15 6 4673 2 63 48
END
これらの行のうち、分子置換法で必要な情報は主に ATOM の行である(HEADER、CRYST1、HETATM の行を残しても問題ない)。また先にも述べたが、PDB ファイルは非対称単位に存在する全ての分子の情報を含んでいるため、複数の分子の情報が含まれている場合も多い。分子置換法では、そのうちのどの分子をモデル分子として選択するかがポイントとなる(補足1)。例えば、1分子のみをモデル分子として使いたい場合には、残りの分子の座標をテキストエディタで削除する(例えば、上に示した赤字の行を削除し END を残す)。そしてそのファイルをセーブした後、COOT で分子を表示し、思惑通りに1分子のみになっているかを確認すると良い。
4. モデル分子の回転/並進
ここでは、3. で準備したモデル分子を回転/並進し、結晶の非対称単位中にモデル分子を当てはめていく操作を実施する。
- Molecular Replacement モジュール内の Molrep – auto MR を選択する。
- Molrep パネルの Data の行の Browse から MTZ file を選択し、Model の行の Browse から準備したモデル分子の pdb file を選択する(図7)。
- 非対称単位中の分子数が判っていれば、Search Options の Number of copies to find に非対称単位中の分子数を入力する(ただし、初期設定で分子を複数当てはめる設定となっているため、不明であれば入力する必要はない)。
- Run→Run Now を選択する。
- CCP4i メインパネルの JOB が FINISHED となっているのを確認後、その JOB をダブルクリックし、Log File タブ内のほぼ最後にある
Summaryを確認する(図8)。以下のような結果が示される。
--- Summary (V0) ---
+------------------------------------------------------------------------------+
| RF TF theta phi chi tx ty tz TF/sg wRfac Score |
+------------------------------------------------------------------------------+
| 1 1 1 146.22 135.90 4.21 0.000 0.000 0.281 12.51 0.531 0.60353|
| 2 2 1 180.00 0.00 3.57 0.504 0.000 0.282 8.40 0.572 0.52812|
| 3 3 10 43.22 -92.26 175.36 0.870 0.000 0.278 1.48 0.636 0.39007|
| 4 4 2 33.93 1.03 157.09 0.622 0.000 0.292 1.63 0.637 0.38946|
| 5 5 2 36.02 -1.64 176.28 0.478 0.000 0.297 1.65 0.638 0.38703|
| 6 12 1 71.43 154.12 49.37 0.977 0.000 0.278 1.85 0.640 0.38378|
| 7 6 6 139.35 -164.51 113.89 0.240 0.000 0.220 1.54 0.640 0.38322|
| 8 11 4 70.65 140.30 36.15 0.326 0.000 0.278 1.56 0.643 0.37775|
| 9 10 6 2.34 17.99 117.27 0.000 0.000 0.223 1.31 0.643 0.37635|
| 10 8 11 156.70 121.38 81.43 0.566 0.000 0.306 1.44 0.642 0.37616|
| 11 7 5 42.77 -176.14 136.31 0.597 0.000 0.329 1.60 0.636 0.37598|
| 12 9 3 87.21 -140.37 70.30 0.329 0.000 0.250 1.35 0.648 0.36549|
+------------------------------------------------------------------------------+
この Summary には、分子を回転(theta, phi, chi)、並進して(tx, ty, tz)した際の TF/sg、wRfac、Score といった情報が示されている。一般に正しい解が得られたとき、TF/sg と Score が他の解に比べて有意に高く、wRfac が有意に低くなる。この例では、一番上の行の解が正しい解であると予想される。
そして Log File のほぼ最後の行には、Molrep が選択した解が以下のように表示される。
Nmon RF TF theta phi chi tx ty tz TF/sg wRfac Score
1 1 1 146.22 135.90 4.21 0.000 0.000 0.281 12.51 0.531 0.603
モデル分子との同一性にもよるが、正しい解が得られている場合に、wRfac は50%台になることが多い。ここで有意な解が得られない場合には、モデル分子の変更(補足1)、X 線回折強度データの質の確認(補足2)、分解能の変更(補足3)などを検討すると良い。また、X 線結晶構造解析を始めたばかりのときには思わぬ落とし穴にはまることもある(補足4)。ここに書いたことを一通り試しても解が得られない場合には、X 線結晶構造解析を複数回経験したことのある研究者に相談してみると良い。
5. 結晶中の分子のパッキングの確認
得られた解の妥当性を評価するために、結晶中の分子のパッキングを確認する。
- CCP4i メインパネルからその JOB をダブルクリックし、Results タブ内の Output Files から Coot を選択する(図9左)。Coot が起動した後、Nomenclature error などが出る場合には Yes を選択して、分子置換後のタンパク質分子を表示する。
- Coot ウインドウ内の Display Manager を選択し、右のカラムの Bonds (Colour by Atom) を C-alphas/Backbone に変更し、Close を選択して Display Manager を閉じる。
- Coot ウインドウ内の Draw から Cell & Symmetry… を選択し、Master Switch: Symmetry On、Symmetry by Molecule… から Display as CAs を選ぶ。Symmetry Atom Display Radius: Radius を30~50程度に設定し、Apply を選択すると、分子の周りに対称操作で関係づけられた分子が表示される(図9右)。
結晶は固体であるので、結晶内では分子同士が接触しているはずである。したがって、上述の操作で隣接する分子を表示させた際に、分子同士が接触しているかどうかを確認する。また、分子が存在しない大きな空間が存在する場合には、その空間に分子が存在する可能性も検討する(補足5)。一方で、この時点で分子同士が重なってしまっている場合には、解が妥当でない可能性がある。そうした場合には、モデル分子の変更(補足1)、X 線回折強度データの質の確認(補足2)、分解能の変更(補足3)を検討し、4. の操作をもう一度行うことをお薦めする。
分子置換法の操作①~⑤を実施した後、CCP4i メインパネルから Refinement モジュールを選択し、Run Refmac5 を選択し、rigid body refinement に続いて、restraint refinement を実施して、構造精密化の段階へと進む。
なお、CCP4 Program Suite 内の分子置換法のソフトとして、Molrep 以外にも Phaser (4), MrBUMP (5), Balbes (6) などが準備されていて、それぞれ特徴があるようである。詳細は Web などで確認して頂き、Molrep でうまく解が見つからない場合にはそれらのソフトの使用も検討すると良い。
以上、CCP4 Program Suite の Molrep の手順を追いながら分子置換法の基礎について述べてきたが、最近では、CCP4 に加えて Phenix (7) の利用者も増えていると思われる。Phenix の場合、X 線回折強度データの評価を phenix.xtriage、分子置換法による位相決定を Phaser-MR、構造精密化を phenix.refine で行う。Phenix には豊富な Tutorial Video があるので、例えば、Checking data quality with phenix.xtriage、How to run Phaser-MR、Phenix tutorial: phenix.refine using default values (GUI) などの動画を参考にしていただくと良いと思う。
工夫とコツ
(補足1)モデル分子の変更
筆者は、図10に示すような D2 対称(分子の中心に222対称)を有する4量体の構造解析を分子置換法で行った際、単量体(水色の分子)、2量体(赤枠、青枠で示した2通り)、および4量体全体をそれぞれモデル分子として用いて、分子置換法を試した。このうち正しい解を得たものは、唯一、赤枠で示した2量体のみであった。このように、多量体を形成しているタンパク質については、複数のモデル分子の可能性を試すことをお薦めする。
また、解が得られない場合には、ポリアラニンモデルをモデル分子として用いてもよい。具体的には、Molrep パネルの Model から Model modification: Polyalanine model を選択すれば良い。また、目的タンパク質とモデル分子とのアミノ酸アライメントも確認し、例えば、保存性の低い領域をテキストエディタで削除したものをモデル分子として使用するなど工夫してみると良い。全て自動処理に任せるのではなく、色々と考えながらモデル分子を選択すると解が得られる場合がある。
(補足2)X 線回折強度データの質の確認
アミノ酸の同一性が高い(例えば50%以上)にもかかわらず、有意な解が得られない場合は、X 線回折強度データそのものの質をチェックすることも重要である。データ全体の CC(1/2) や R-merge(R-FACTOR observed)をチェックすると良い(図11)。X 線回折強度データ処理ソフト XDS で処理した場合には、CORRECT.LP あるいは XSCALE.LP というファイルをメモ帳などのテキストエディタで開き、最終行に移動してから「-3.0」を「上へ」で検索すれば統計データが確認できる。
また、X 線回折強度データ処理の段階では、空間群が決定できない場合も多い。そうした場合には、無理に一つの空間群に決定する必要はない。可能性のある全ての空間群(厳密には Laue 対称)を用いて XDS でそれぞれ処理し、それぞれの X 線回折強度データを用いて分子置換を行うと良い。
(補足3)分解能の変更
分子置換法を行うにあたり、分解能を変更すると良い。例えば、2.0 Å 分解能の X 線回折強度データが得られている場合、筆者は、2.0 Å, 3.0 Å, 4.0 Å, 6.0 Å のように最高分解能を変えて分子置換法を実施する場合も多い。また、そうした場合に、ある条件のときのみ、正しい解が得られることもあるので試して頂きたい。具体的には、Molrep パネルの Experimental Data 内の High resolution cut-off にチェックを入れて、最高分解能の値を入力すれば良い。ただし、分子置換法で解を得た後の Rigid body refinement や構造精密化では、全てのデータ(この場合は 2.0 Å 分解能)を用いる必要がある。
(補足4)思わぬ落とし穴にはまった例
アミノ酸同一性が高いモデル分子があるにもかかわらず、どうしても解が見つからないと学生さんが言ってきたので、一緒に調べてみたところ、少し難しいケースであったので参考までにここに示す。
まず、学生さんが持ってきた X 線回折強度データは X 線回折強度データ処理ソフト XDS を使って空間群 P222 で処理されたものであった。これは、回折 X 線強度の対称である Laue 対称を mmm として処理したデータであることを意味し、P222 は仮の空間群である。また厄介なことに補足1で述べたように可能性のあるモデル分子が複数あった。そこで、可能性のあるモデル分子を全て準備して、それぞれのモデル分子につき Molrep パネルの Search Options から SG to use: Laue class を選択することで、モデル分子と空間群の候補をすべて試してみた。その結果、図12左に示したように、Molrep のログファイルにて高い Score 値と Contrast 値を示すモデル分子と空間群の組み合わせが見つかった(ここでは P22121 という空間群が候補)。しかし、P22121 という空間群は厳密には存在しない(詳細は、空間群の詳細が記載されている書籍等をご参照いただきたい)。そこで、CCP4i の Reindex を用いて空間群を P21212 に変換した後、その MTZ file を用いてもう一度 Molrep を流して、ようやく正しい解を得ることができた(図12右)。こうした少し難しいケースもあるので、構造解析を複数回経験したことのある研究者に相談してみると良いこともある。
(補足5)結晶中の分子のパッキング
分子置換法では、一般に、非対称単位に当てはめる分子の数が多いほど、正しい解を得ることは困難となり、ソフトで全ての分子を見つけることができない場合もある。そうした場合には、一部をマニュアルで当てはめることを検討すると良い。例えば、図13のように、ソフトで8分子中7分子が見つかったが、8分子目の解がどうしても見つからなかったケースがあった。ここで、分子のパッキングを確認したところ、もう1つの分子が存在すると予想される空間が存在した。このとき、タンパク質は2量体構造をとっていたために、青色の分子と2量体を形成するようにもう1つの分子をマニュアルで当てはめて、構造解析できたケースもあった。
文献
- Winn, MD, et al., Acta Cryst. D67, 235–242 (2011)
- Vagin, A, et al., J. Appl. Cryst. 30, 1022–1025 (1997)
- Matthews, BW, J. Mol. Biol. 33, 491–497 (1968)
- McCoy, J, et al., J. Appl. Cryst. 40, 658–764 (2007)
- Keegan, RM and Winn, MD, Acta Cryst. D63, 447–457 (2007)
- Long, F, et al., Acta Cryst. D64, 125–132 (2008)
- Liebschner, D, et al., Acta Cryst. D75, 861–877 (2019)
-
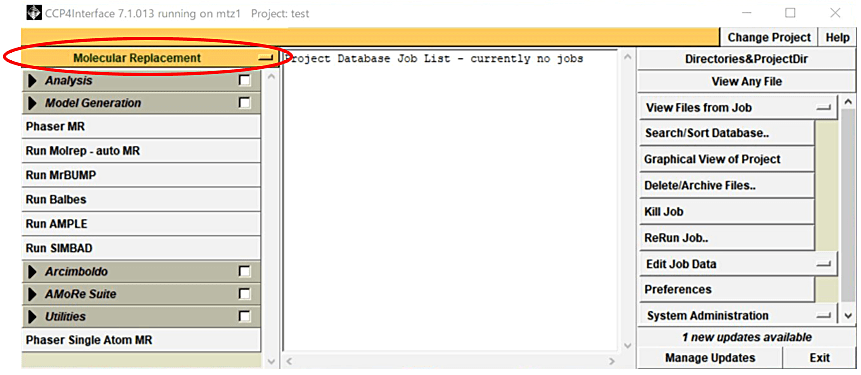
図1:CCP4i のメインパネルから Molecular Replacement モジュール(赤丸)を選択する。 -
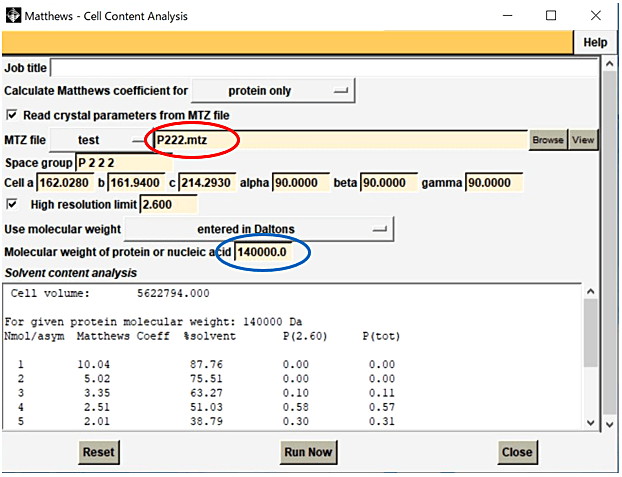
図2:Matthews - Cell Content Analysis パネルで MTZ file を Browse から選択し(赤丸)、おおよその分子量を入力し(青丸)、Run Now を選択する(注:図2、図3、図7で異なる MTZ file 名となっているが、実際は同じファイルを使用する必要がある)。 -
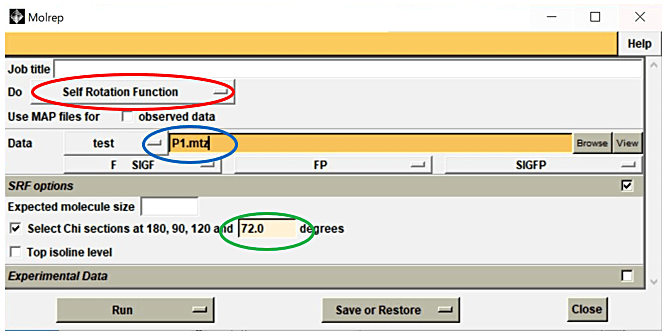
図3:Molrep パネルにて、Do Self rotation function を選択し(赤丸)、MTZ file を Browse から選択する(青丸)。必要であれば、SRF options 内の Select Chi sections を選択して、NCS の候補の角度を入力し(緑丸)、Run→Run Now を選択する。 -
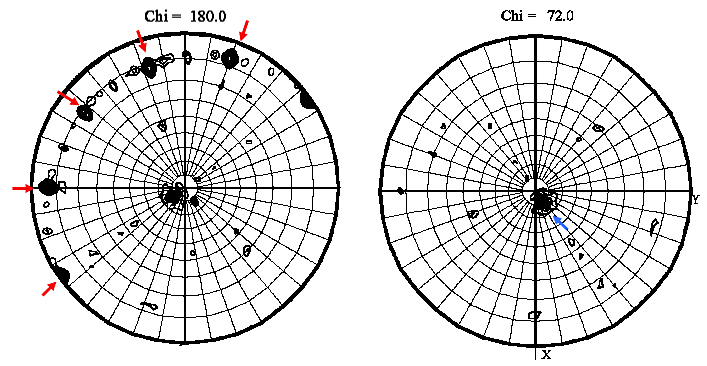
図4:極座標で示された自己回転関数の例。 -
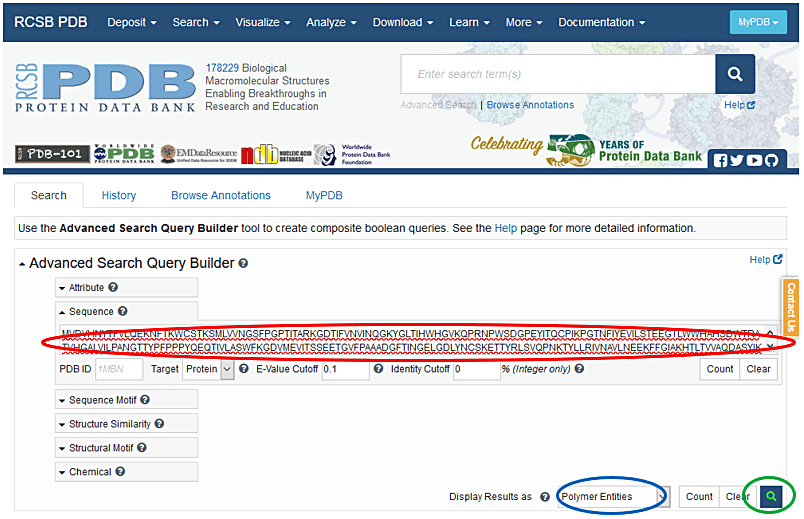
図5:Protein Data Bank で Search→Sequence Search→Advanced Search - Sequence Search を選択し、アミノ酸配列(赤丸)、Polymer Entities を選択し(青丸)、虫眼鏡のマークを選択する(緑丸)。 -
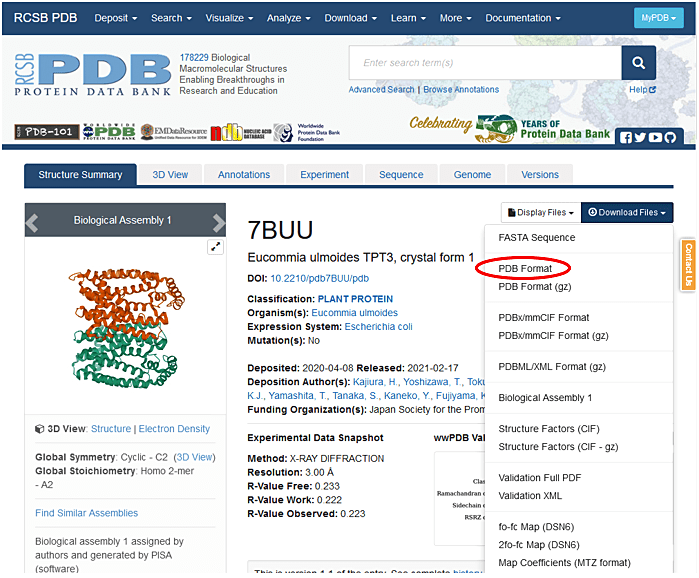
図6:Protein Data Bank で、PDB ID を選択した後、Download Files から PDB Format を選択して PDB ファイルをダウンロードする(赤丸)。 -
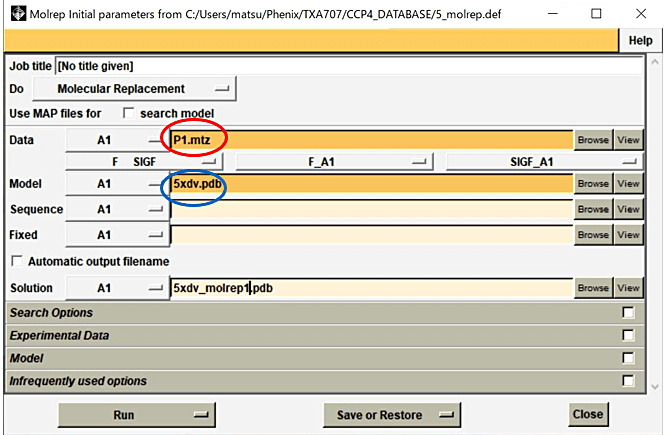
図7:Molrep パネルの選択画面。この画面で Data の行の Browse から MTZ file を選択し(赤丸)、Mode の行の Browse から準備したモデル分子の pdb file を選択した後(青丸)、Run→Run Now を選択する。 -
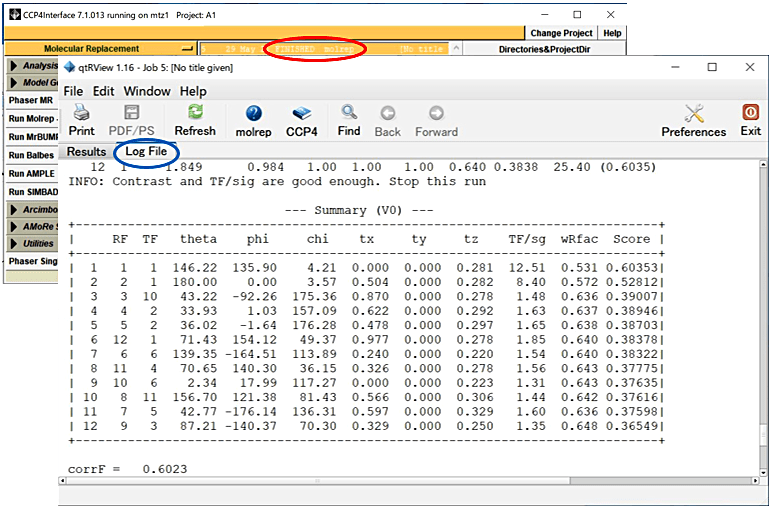
図8:CCP4i メインパネルの JOB が FINISHED となっているのを確認後(赤丸)、その JOB(赤丸部分)をダブルクリックし、Log File タブ(青丸)内のほぼ最後にある Summary を確認する。 -
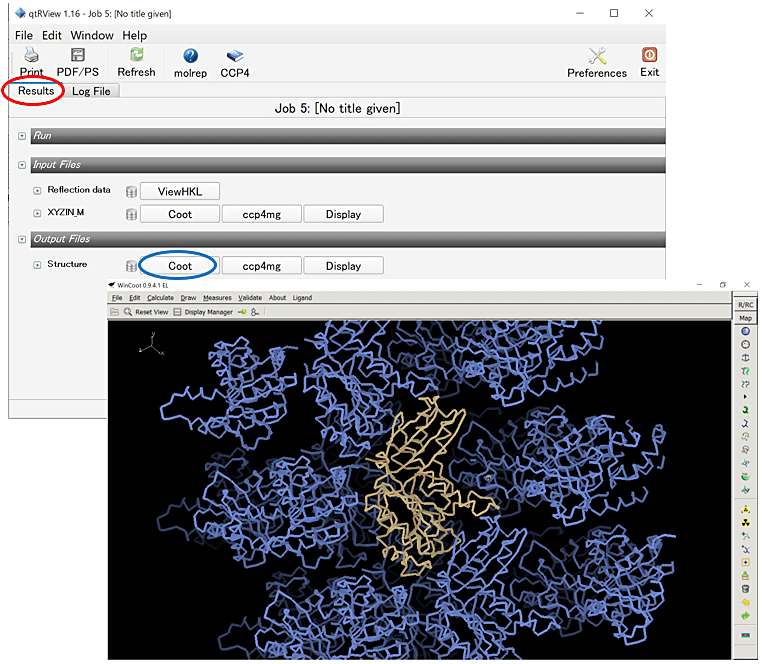
図9:CCP4i メインパネルから JOB をダブルクリックし、Results タブ(赤丸)内の Output Files から Coot(青丸)を選択する。5. の①~③を実施すれば、右下の図のように結晶中の分子のパッキングが表示される。 -
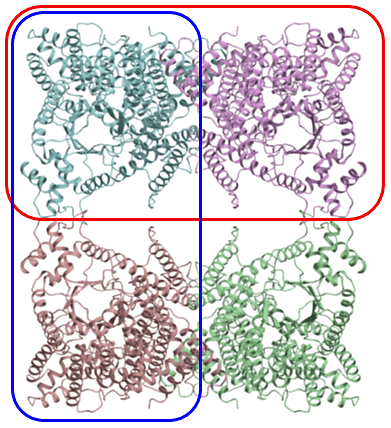
図10:分子内に D2 対称(中心に222対称)がある4量体タンパク質の例。 -
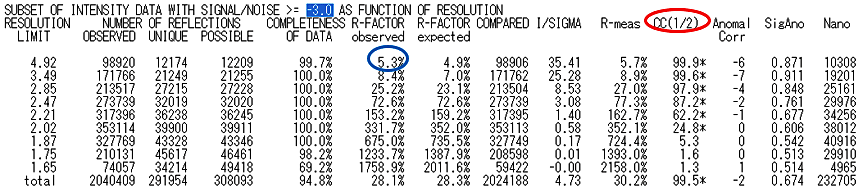
図11:XDS の CORRECT.LP 内に書かれている統計テーブルの例。赤丸で示した CC(1/2) は60%以上の分解能が目安と思われるので、この図のデータでは最高分解能は 2.2 Å 程度を選択すべきである。また、青丸で示した低角範囲の R-FACTOR observed(R-merge)が10%を超えるような高い値を示す場合には、データそのものが悪い可能性や空間群が間違っている可能性もある(その点に関してこの図のデータでは大きな問題はなさそうである)。 -
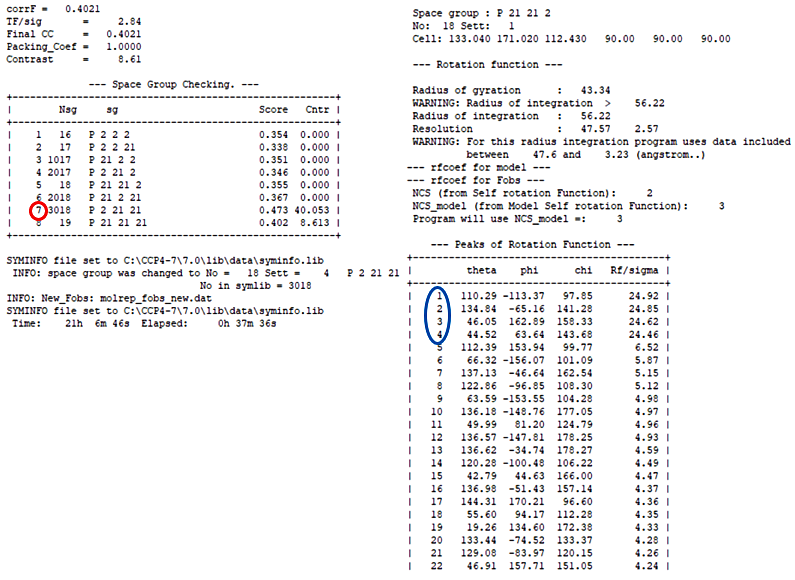
図12:Molrep のログファイルの例。左図において、高い Score 値と Contrast 値を示す空間群(Space group、ここでは sg と示している)は P22121 であったが(赤丸)、P22121 という空間群は厳密には存在しないため、これを P21212 に変更する必要があった。そこで、CCP4i の Reindex を用いて空間群を P21212 に変更した後、その MTZ file を用いてもう一度 Molrep を流すことで正しい解が得られた(青丸の4つ解が正しい解であった)。 -
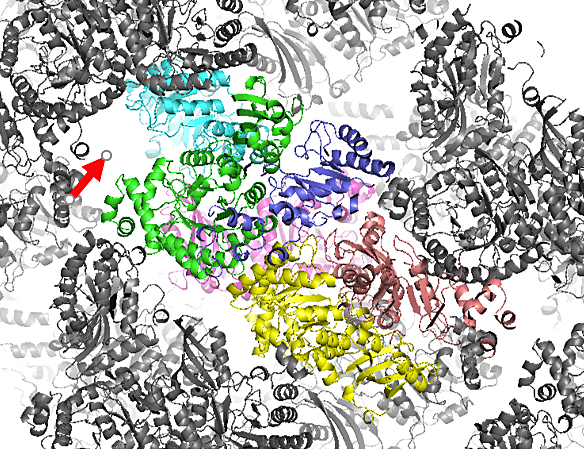
図13:結晶中の分子のパッキングの例。8分子中7分子が分子置換法で見つかったが、分子のパッキングを確認したところ、赤矢印に示した空間が存在し、もう1つの分子が存在すると予想された。そこで、水色の分子と2量体を形成するようにもう1つの分子をマニュアルで当てはめて構造解析することができた。
概要
X 線結晶構造解析法によってタンパク質を構造解析するためには、まずそのタンパク質を結晶化する必要がある。そして結晶が得られれば、その結晶を用いて X 線回折実験を行い、X 線回折強度データを収集する。そこで得られた X 線回折強度データを用いて構造解析を行うが、この際に位相情報が必要となる。というのも、X 線回折強度データには構造解析に必要な位相情報が含まれないからである(詳細は、「X 線、位相問題」で検索し調べていただきたい)。その位相情報を得る手段として、分子置換法(MR 法)、重原子同型置換法(MIR 法)、多波長/単波長異常分散法(MAD/SAD 法)といった方法が用いられる。なかでも、類似のタンパク質の立体構造を使って位相情報を得る分子置換法はまず初めに試すべき方法であろう。なぜなら、2021年5月の時点で、Protein Data Bank Japan(https://pdbj.org/)には17万を超えるタンパク質の立体構造情報(PDB データ)が登録されているからで、今後構造解析されるタンパク質の多くは、類似のタンパク質の立体構造が登録されている可能性が高いからである。
さて、分子置換法は、1. 非対称単位中のタンパク質分子数の見積もり、2. 自己回転関数による非結晶学的回転軸の確認、3. 類似タンパク質(モデル分子)の選択と編集、4. モデル分子の回転/並進、5. 結晶中の分子のパッキングの確認という5段階に分けられる。これらは、CCP4 Program Suite (1) が導入されている Windows や Mac のパソコンさえあれば、1日で実施・完了できる。ただし、類似タンパク質とのアミノ酸同一性が低い場合(例えば20%以下の場合)には、4. の段階で容易に結果が得られずより多くの時間がかかることをご承知おき頂きたい。
ここでは初めて構造解析を実施する研究者を対象として、CCP4 Program Suite が導入されている Windows のパソコンを用いて、CCP4 Program Suite の Molrep (2) という分子置換法のソフトを用いた手順を解説する。
装置
CCP4 Program Suite(2021年5月現在 v7.1.013)と COOT が導入されている Windows パソコンを準備する。なお、Windows パソコンに CCP4 と COOT を導入するためには、「CCP4 Windows」で検索し、「CCP4 Program Suite including SHELX and COOT」をダウンロードした後、そのファイルをダブルクリックして「詳細」 を選択した後に実行するとよい。
実験手順
- 非対称単位中のタンパク質分子数の見積もり
- 自己回転関数による非結晶学的回転軸の確認
- 類似タンパク質(モデル分子)の選択と編集
- モデル分子の回転/並進
- 結晶中の分子のパッキングの確認
実験の詳細
本プロトコールを始める前に
目的タンパク質のアミノ酸配列が既知であり、X 線回折強度データファイル(MTZ File)が得られていることを前提として手順を解説する。
分子置換法の詳細
1. 非対称単位中のタンパク質分子数の見積もり
非対称単位とは、結晶中で行う対称操作により重ねることができない最小の領域のことを指す。なお、Protein Data Bank に登録されている立体構造情報は、通常は非対称単位に存在する全ての分子の情報を含んでいる。
非対称単位は、例えば空間群が P21 の場合には単位胞の 1/2 の大きさを持ち、空間群 が P43212 の場合には単位胞の 1/8 となる。このように空間群によって単位胞に対する非対称単位の割合が異なるので、詳しく知りたい場合には空間群の詳細が記載されている書籍(例えば、International Tables for Crystallography, Volume A, ISBN: 978-0-470-97423-0)をご参照いただきたい。
さて、分子置換法とは、類似タンパク質を回転・並進させ、非対称単位に存在する目的分子に対して類似タンパク質を重ねる操作法のことである。そのとき、非対称単位内に複数の分子が存在すれば、その分子全てに対して類似タンパク質を重ねる必要がある。したがって、まず非対称単位に何分子が含まれているかを以下の手順で見積もることから始める。
- CCP4i を起動し、Project 名と X 線回折強度データファイル(MTZ File)が存在するフォルダ設定した後に、CCP4i のメインパネルから Molecular Replacement モジュールを選択する(図1)。
- Analysis → Cell Content Analysis を選択すると Matthews - Cell Content Analysis パネルが起動する(図2)。
- Matthews ‒ Cell Content Analysis のパネルにて、MTZ file を Browse から選択すると、空間群、格子定数、分解能が自動で表示される。
- Molecular weight of protein or nucleic acid の欄に分子量を入力する。
- Run Now を選択する。
以下のような結果が表示される(以下は一例)。
Nmol/asym Matthews Coeff %solvent P(2.60) P(tot)
1 10.04 87.76 0.00 0.00
2 5.02 75.51 0.00 0.00
3 3.35 63.27 0.10 0.11
4 2.51 51.03 0.58 0.57
5 2.01 38.79 0.30 0.31
6 1.67 26.54 0.00 0.00
7 1.43 14.30 0.00 0.00
8 1.26 2.06 0.00 0.00
通常、表の Matthews Coeff で示されている値は 1.7~3.5 A3/Da 程度とされるため (3)、この場合は、非対称単位中におおよそ3~5分子が存在すると予想される。この段階では、分子の数を厳密に決定する必要はなく、ある程度の可能性を検討できれば、次の段階に進む。
2. 自己回転関数による非結晶学的回転軸の確認
非対称単位に複数の分子が存在することがわかれば、次に自己回転関数を計算し、非結晶学的回転軸(NCS)の存在を確認する。
- Molecular Replacement モジュール内の Molrep – auto MR を選択する。
- Molrep パネルにて、Do Self rotation function を選択し、MTZ file を Browse から選択する(図3)。
- 必要であれば、SRF options 内の Select Chi sections を選択して、NCS の候補の角度を入力する。例えば、2量体が5つ集まった10量体が存在している分子では、分子内に2回軸が5本と5回軸が存在する。初期値で180度は設定されているので、設定されていない5回軸(360度を5で割った数字72)を Select Chi sections に入力すると良い。
- Run→Run Now を選択する。
1. の①で指定したフォルダに .ps という拡張子のついた Postscript ファイルが出力される。このファイルを Adobe Distiller で PDF ファイルに変換した後に開くと(Adobe Illustrator ではそのまま閲覧可能)、極座標で示された図が表示される(図4)。
図4では、2回軸の存在を示す Chi = 180 のセクションに5つの高いピーク(上図の赤矢印)が等間隔で存在しており、Chi = 72 のセクションに1つの高いピーク(上図の青矢印)が存在していることから、2量体が5つ集まった10量体が存在している可能性が予想される。この自己回転関数の計算結果と、1. で見積もった非対称単位内の分子数とを比較して、結晶中で分子が何分子どのように配置しているかを予想しておく。
ただし、この 2. は分子内に NCS が存在して、なおかつそれが結晶学的回転軸とは異なる場合にのみ有用となる。そのため、図4のような明解な情報が得られない場合も多く、その場合は気にせず次の段階に進む。
3. 類似タンパク質(モデル分子)の選択と編集
まず、Protein Data Bank に登録されているタンパク質の中で、目的タンパク質とアミノ酸同一性の高いタンパク質(モデル分子)を選択する。モデル分子の選択は様々な方法があるが、以下では、Protein Data Bank から同一性の高いものを選択する方法を示す。
- https://www.rcsb.org/ にアクセスし Search→Sequence Search→Advanced Search - Sequence Search を選択する(図5)。
- Sequence? の空欄にアミノ酸配列(一文字記号)を入力する。
- Display Results as で Polymer Entities を選択し、虫眼鏡のマークを選択する。
- Sequence identity と E-value を確認する。私見であるが Sequence identity が20%以上あれば分子置換法を試す価値はあると思われる。Download Alignment を選択して Alignment ファイルをダウンロードしておく。その Alignment ファイルは、CLUSTALX というソフト等で表示できる。
- Sequence identity が高く E-value が小さい PDB ID を選択し、Download Files から PDB Format を選択して PDB ファイルをダウンロードし(図6)、そのファイルを 1. の①で指定したフォルダに保存する。
次に、テキストエディタ(例えば、Windows アクセサリのメモ帳)で PDB ファイルを開いてファイルの内容を確認する。PDB ファイルの主な行を説明すると、HEADER で始まる行には PDB ID が示され、CRYST1 には単位格子と空間群の情報が書かれている。ATOM にはタンパク質の座標データが記され、HETATM にはタンパク質に結合している基質アナログや水分子などの座標データが書かれている。
HEADER CELL CYCLE 05-NOV-16 5H5G
TITLE STAPHYLOCOCCUS AUREUS FTSZ-GDP IN T AND R STATES
COMPND MOL_ID: 1;
COMPND 2 MOLECULE: CELL DIVISION PROTEIN FTSZ;
(略)
CRYST1 43.917 159.023 44.058 90.00 92.78 90.00 P 1 21 1 4
ORIGX1 1.000000 0.000000 0.000000 0.00000
ORIGX2 0.000000 1.000000 0.000000 0.00000
ORIGX3 0.000000 0.000000 1.000000 0.00000
SCALE1 0.022770 0.000000 0.001104 0.00000
SCALE2 0.000000 0.006288 0.000000 0.00000
SCALE3 0.000000 0.000000 0.022724 0.00000
ATOM 1 N MET A 11 70.851 24.422 76.500 1.00 58.77 N
ATOM 2 CA MET A 11 69.861 23.493 75.871 1.00 60.16 C
ATOM 3 C MET A 11 70.402 23.013 74.526 1.00 54.98 C
ATOM 4 O MET A 11 70.878 23.819 73.724 1.00 52.56 O
ATOM 5 CB MET A 11 68.489 24.170 75.715 1.00 65.01 C
ATOM 6 CG MET A 11 67.383 23.242 75.219 1.00 70.22 C
ATOM 7 SD MET A 11 65.810 23.448 76.090 1.00 79.56 S
ATOM 8 CE MET A 11 64.628 22.796 74.912 1.00 76.98 C
ATOM 9 N ALA A 12 70.336 21.703 74.294 1.00 51.77 N
ATOM 10 CA ALA A 12 71.092 21.069 73.206 1.00 46.93 C
ATOM 11 C ALA A 12 70.521 21.394 71.823 1.00 42.06 C
ATOM 12 O ALA A 12 69.319 21.313 71.612 1.00 42.26 O
ATOM 13 CB ALA A 12 71.164 19.568 73.419 1.00 47.58 C
(略)
TER
ATOM 2229 N HIS B 10 30.445 -18.763 57.180 1.00 84.67 N
ATOM 2230 CA HIS B 10 29.963 -18.655 55.760 1.00 88.20 C
ATOM 2231 C HIS B 10 31.005 -19.137 54.736 1.00 83.06 C
ATOM 2232 O HIS B 10 30.737 -20.057 53.965 1.00 80.48 O
ATOM 2233 CB HIS B 10 29.535 -17.216 55.446 1.00 89.27 C
ATOM 2234 CG HIS B 10 29.024 -17.028 54.051 1.00 92.35 C
ATOM 2235 ND1 HIS B 10 27.735 -17.348 53.681 1.00 94.12 N
ATOM 2236 CD2 HIS B 10 29.628 -16.553 52.937 1.00 93.17 C
ATOM 2237 CE1 HIS B 10 27.565 -17.076 52.400 1.00 93.87 C
ATOM 2238 NE2 HIS B 10 28.699 -16.591 51.925 1.00 96.73 N
ATOM 2239 N MET B 11 32.176 -18.498 54.738 1.00 78.54 N
ATOM 2240 CA MET B 11 33.291 -18.831 53.841 1.00 75.35 C
ATOM 2241 C MET B 11 34.497 -19.169 54.713 1.00 66.55 C
ATOM 2242 O MET B 11 34.821 -18.415 55.632 1.00 67.02 O
ATOM 2243 CB MET B 11 33.608 -17.626 52.948 1.00 80.23 C
ATOM 2244 CG MET B 11 34.364 -17.949 51.667 1.00 84.89 C
ATOM 2245 SD MET B 11 33.314 -18.347 50.247 1.00 90.91 S
ATOM 2246 CE MET B 11 32.482 -16.780 49.969 1.00 92.25 C
(略)
TER 4475 PHE B 315
HETATM 4476 PB GDP A 401 73.156 10.830 45.052 1.00 29.42 P
HETATM 4477 O1B GDP A 401 74.218 10.572 44.048 1.00 33.95 O
HETATM 4478 O2B GDP A 401 72.385 12.106 44.954 1.00 30.71 O
HETATM 4479 O3B GDP A 401 73.676 10.647 46.448 1.00 36.84 O
HETATM 4480 O3A GDP A 401 72.122 9.629 44.803 1.00 30.55 O
G(略)
MASTER 319 0 3 26 23 0 15 6 4673 2 63 48
END
これらの行のうち、分子置換法で必要な情報は主に ATOM の行である(HEADER、CRYST1、HETATM の行を残しても問題ない)。また先にも述べたが、PDB ファイルは非対称単位に存在する全ての分子の情報を含んでいるため、複数の分子の情報が含まれている場合も多い。分子置換法では、そのうちのどの分子をモデル分子として選択するかがポイントとなる(補足1)。例えば、1分子のみをモデル分子として使いたい場合には、残りの分子の座標をテキストエディタで削除する(例えば、上に示した赤字の行を削除し END を残す)。そしてそのファイルをセーブした後、COOT で分子を表示し、思惑通りに1分子のみになっているかを確認すると良い。
4. モデル分子の回転/並進
ここでは、3. で準備したモデル分子を回転/並進し、結晶の非対称単位中にモデル分子を当てはめていく操作を実施する。
- Molecular Replacement モジュール内の Molrep – auto MR を選択する。
- Molrep パネルの Data の行の Browse から MTZ file を選択し、Model の行の Browse から準備したモデル分子の pdb file を選択する(図7)。
- 非対称単位中の分子数が判っていれば、Search Options の Number of copies to find に非対称単位中の分子数を入力する(ただし、初期設定で分子を複数当てはめる設定となっているため、不明であれば入力する必要はない)。
- Run→Run Now を選択する。
- CCP4i メインパネルの JOB が FINISHED となっているのを確認後、その JOB をダブルクリックし、Log File タブ内のほぼ最後にある
Summaryを確認する(図8)。以下のような結果が示される。
--- Summary (V0) ---
+------------------------------------------------------------------------------+
| RF TF theta phi chi tx ty tz TF/sg wRfac Score |
+------------------------------------------------------------------------------+
| 1 1 1 146.22 135.90 4.21 0.000 0.000 0.281 12.51 0.531 0.60353|
| 2 2 1 180.00 0.00 3.57 0.504 0.000 0.282 8.40 0.572 0.52812|
| 3 3 10 43.22 -92.26 175.36 0.870 0.000 0.278 1.48 0.636 0.39007|
| 4 4 2 33.93 1.03 157.09 0.622 0.000 0.292 1.63 0.637 0.38946|
| 5 5 2 36.02 -1.64 176.28 0.478 0.000 0.297 1.65 0.638 0.38703|
| 6 12 1 71.43 154.12 49.37 0.977 0.000 0.278 1.85 0.640 0.38378|
| 7 6 6 139.35 -164.51 113.89 0.240 0.000 0.220 1.54 0.640 0.38322|
| 8 11 4 70.65 140.30 36.15 0.326 0.000 0.278 1.56 0.643 0.37775|
| 9 10 6 2.34 17.99 117.27 0.000 0.000 0.223 1.31 0.643 0.37635|
| 10 8 11 156.70 121.38 81.43 0.566 0.000 0.306 1.44 0.642 0.37616|
| 11 7 5 42.77 -176.14 136.31 0.597 0.000 0.329 1.60 0.636 0.37598|
| 12 9 3 87.21 -140.37 70.30 0.329 0.000 0.250 1.35 0.648 0.36549|
+------------------------------------------------------------------------------+
この Summary には、分子を回転(theta, phi, chi)、並進して(tx, ty, tz)した際の TF/sg、wRfac、Score といった情報が示されている。一般に正しい解が得られたとき、TF/sg と Score が他の解に比べて有意に高く、wRfac が有意に低くなる。この例では、一番上の行の解が正しい解であると予想される。
そして Log File のほぼ最後の行には、Molrep が選択した解が以下のように表示される。
Nmon RF TF theta phi chi tx ty tz TF/sg wRfac Score
1 1 1 146.22 135.90 4.21 0.000 0.000 0.281 12.51 0.531 0.603
モデル分子との同一性にもよるが、正しい解が得られている場合に、wRfac は50%台になることが多い。ここで有意な解が得られない場合には、モデル分子の変更(補足1)、X 線回折強度データの質の確認(補足2)、分解能の変更(補足3)などを検討すると良い。また、X 線結晶構造解析を始めたばかりのときには思わぬ落とし穴にはまることもある(補足4)。ここに書いたことを一通り試しても解が得られない場合には、X 線結晶構造解析を複数回経験したことのある研究者に相談してみると良い。
5. 結晶中の分子のパッキングの確認
得られた解の妥当性を評価するために、結晶中の分子のパッキングを確認する。
- CCP4i メインパネルからその JOB をダブルクリックし、Results タブ内の Output Files から Coot を選択する(図9左)。Coot が起動した後、Nomenclature error などが出る場合には Yes を選択して、分子置換後のタンパク質分子を表示する。
- Coot ウインドウ内の Display Manager を選択し、右のカラムの Bonds (Colour by Atom) を C-alphas/Backbone に変更し、Close を選択して Display Manager を閉じる。
- Coot ウインドウ内の Draw から Cell & Symmetry… を選択し、Master Switch: Symmetry On、Symmetry by Molecule… から Display as CAs を選ぶ。Symmetry Atom Display Radius: Radius を30~50程度に設定し、Apply を選択すると、分子の周りに対称操作で関係づけられた分子が表示される(図9右)。
結晶は固体であるので、結晶内では分子同士が接触しているはずである。したがって、上述の操作で隣接する分子を表示させた際に、分子同士が接触しているかどうかを確認する。また、分子が存在しない大きな空間が存在する場合には、その空間に分子が存在する可能性も検討する(補足5)。一方で、この時点で分子同士が重なってしまっている場合には、解が妥当でない可能性がある。そうした場合には、モデル分子の変更(補足1)、X 線回折強度データの質の確認(補足2)、分解能の変更(補足3)を検討し、4. の操作をもう一度行うことをお薦めする。
分子置換法の操作①~⑤を実施した後、CCP4i メインパネルから Refinement モジュールを選択し、Run Refmac5 を選択し、rigid body refinement に続いて、restraint refinement を実施して、構造精密化の段階へと進む。
なお、CCP4 Program Suite 内の分子置換法のソフトとして、Molrep 以外にも Phaser (4), MrBUMP (5), Balbes (6) などが準備されていて、それぞれ特徴があるようである。詳細は Web などで確認して頂き、Molrep でうまく解が見つからない場合にはそれらのソフトの使用も検討すると良い。
以上、CCP4 Program Suite の Molrep の手順を追いながら分子置換法の基礎について述べてきたが、最近では、CCP4 に加えて Phenix (7) の利用者も増えていると思われる。Phenix の場合、X 線回折強度データの評価を phenix.xtriage、分子置換法による位相決定を Phaser-MR、構造精密化を phenix.refine で行う。Phenix には豊富な Tutorial Video があるので、例えば、Checking data quality with phenix.xtriage、How to run Phaser-MR、Phenix tutorial: phenix.refine using default values (GUI) などの動画を参考にしていただくと良いと思う。
工夫とコツ
(補足1)モデル分子の変更
筆者は、図10に示すような D2 対称(分子の中心に222対称)を有する4量体の構造解析を分子置換法で行った際、単量体(水色の分子)、2量体(赤枠、青枠で示した2通り)、および4量体全体をそれぞれモデル分子として用いて、分子置換法を試した。このうち正しい解を得たものは、唯一、赤枠で示した2量体のみであった。このように、多量体を形成しているタンパク質については、複数のモデル分子の可能性を試すことをお薦めする。
また、解が得られない場合には、ポリアラニンモデルをモデル分子として用いてもよい。具体的には、Molrep パネルの Model から Model modification: Polyalanine model を選択すれば良い。また、目的タンパク質とモデル分子とのアミノ酸アライメントも確認し、例えば、保存性の低い領域をテキストエディタで削除したものをモデル分子として使用するなど工夫してみると良い。全て自動処理に任せるのではなく、色々と考えながらモデル分子を選択すると解が得られる場合がある。
(補足2)X 線回折強度データの質の確認
アミノ酸の同一性が高い(例えば50%以上)にもかかわらず、有意な解が得られない場合は、X 線回折強度データそのものの質をチェックすることも重要である。データ全体の CC(1/2) や R-merge(R-FACTOR observed)をチェックすると良い(図11)。X 線回折強度データ処理ソフト XDS で処理した場合には、CORRECT.LP あるいは XSCALE.LP というファイルをメモ帳などのテキストエディタで開き、最終行に移動してから「-3.0」を「上へ」で検索すれば統計データが確認できる。
また、X 線回折強度データ処理の段階では、空間群が決定できない場合も多い。そうした場合には、無理に一つの空間群に決定する必要はない。可能性のある全ての空間群(厳密には Laue 対称)を用いて XDS でそれぞれ処理し、それぞれの X 線回折強度データを用いて分子置換を行うと良い。
(補足3)分解能の変更
分子置換法を行うにあたり、分解能を変更すると良い。例えば、2.0 Å 分解能の X 線回折強度データが得られている場合、筆者は、2.0 Å, 3.0 Å, 4.0 Å, 6.0 Å のように最高分解能を変えて分子置換法を実施する場合も多い。また、そうした場合に、ある条件のときのみ、正しい解が得られることもあるので試して頂きたい。具体的には、Molrep パネルの Experimental Data 内の High resolution cut-off にチェックを入れて、最高分解能の値を入力すれば良い。ただし、分子置換法で解を得た後の Rigid body refinement や構造精密化では、全てのデータ(この場合は 2.0 Å 分解能)を用いる必要がある。
(補足4)思わぬ落とし穴にはまった例
アミノ酸同一性が高いモデル分子があるにもかかわらず、どうしても解が見つからないと学生さんが言ってきたので、一緒に調べてみたところ、少し難しいケースであったので参考までにここに示す。
まず、学生さんが持ってきた X 線回折強度データは X 線回折強度データ処理ソフト XDS を使って空間群 P222 で処理されたものであった。これは、回折 X 線強度の対称である Laue 対称を mmm として処理したデータであることを意味し、P222 は仮の空間群である。また厄介なことに補足1で述べたように可能性のあるモデル分子が複数あった。そこで、可能性のあるモデル分子を全て準備して、それぞれのモデル分子につき Molrep パネルの Search Options から SG to use: Laue class を選択することで、モデル分子と空間群の候補をすべて試してみた。その結果、図12左に示したように、Molrep のログファイルにて高い Score 値と Contrast 値を示すモデル分子と空間群の組み合わせが見つかった(ここでは P22121 という空間群が候補)。しかし、P22121 という空間群は厳密には存在しない(詳細は、空間群の詳細が記載されている書籍等をご参照いただきたい)。そこで、CCP4i の Reindex を用いて空間群を P21212 に変換した後、その MTZ file を用いてもう一度 Molrep を流して、ようやく正しい解を得ることができた(図12右)。こうした少し難しいケースもあるので、構造解析を複数回経験したことのある研究者に相談してみると良いこともある。
(補足5)結晶中の分子のパッキング
分子置換法では、一般に、非対称単位に当てはめる分子の数が多いほど、正しい解を得ることは困難となり、ソフトで全ての分子を見つけることができない場合もある。そうした場合には、一部をマニュアルで当てはめることを検討すると良い。例えば、図13のように、ソフトで8分子中7分子が見つかったが、8分子目の解がどうしても見つからなかったケースがあった。ここで、分子のパッキングを確認したところ、もう1つの分子が存在すると予想される空間が存在した。このとき、タンパク質は2量体構造をとっていたために、青色の分子と2量体を形成するようにもう1つの分子をマニュアルで当てはめて、構造解析できたケースもあった。
文献
- Winn, MD, et al., Acta Cryst. D67, 235–242 (2011)
- Vagin, A, et al., J. Appl. Cryst. 30, 1022–1025 (1997)
- Matthews, BW, J. Mol. Biol. 33, 491–497 (1968)
- McCoy, J, et al., J. Appl. Cryst. 40, 658–764 (2007)
- Keegan, RM and Winn, MD, Acta Cryst. D63, 447–457 (2007)
- Long, F, et al., Acta Cryst. D64, 125–132 (2008)
- Liebschner, D, et al., Acta Cryst. D75, 861–877 (2019)
