

概要等
核磁気共鳴(NMR)を用いた溶液中での蛋白質立体構造解析では、対象分子のサイズに応じて適切な試料調製法と測定法を選択する必要がある。分子量20 k程度までの蛋白質では、\(\ce{^{13}C}\)/\(\ce{^{15}N}\)均一ラベル試料を用いた解析手法が確立している。本稿では主にこの範囲を対象とする。一方、30 kを超える高分子量蛋白質では、TROSYベースの測定法と重水素化ラベルの導入が必要となる。さらに50 kを超える蛋白質に対しては、分子全体の原子座標という意味での立体構造を求めるのはかなり困難となる。なお、ここでの分子量は溶液中の実効値であり、二量体などの会合体では見かけ上大きくなる点に留意すべきである。また、分子量数千のペプチドに対しては同位体ラベルは必須ではないが、同位体ラベルできれば、自動帰属・構造計算が容易となる。
NMRによる解析手順は大きく二段階に分けられる。第一は化学シフトの帰属であり、各原子核の信号を分子構造中の対応する原子に結び付ける作業である。特に主鎖原子の帰属は比較的容易で、構造解析のみならず相互作用研究にも有用である。近年では自動化手法の導入により効率化が進んでいる。第二は、核オーバーハウザー効果(NOE)などの距離情報や角度制約を用いた構造計算である。また、化学シフト値自体を利用した二次構造予測や動的解析も一般的である。分子量50 k超の蛋白質であっても、メチル基特異的標識を組み合わせることで、部分的な構造情報を得ることは有益であり、NMRは単なる構造決定手段にとどまらず、機能解明に直結する情報源としての役割を拡大している。
なお、NMRの測定法や原理については優れた書籍が多数あるが、蛋白質に関しては、書籍(1)が測定の理論的側面から詳しく記述されており大変参考になる。また書籍(2)は蛋白質に関するものでは無いが、実験の実際的側面と基本的原理が分かり易く述べられている。
装置
- \(\ce{^{1}H}\)/\(\ce{^{13}C}\)/\(\ce{^{15}N}\)三重共鳴プローブが付いた三重共鳴実験可能な500 MHz以上のNMR装置
- 解析ソフトウエアがインストールされているPC
実験手順
- 準備
- 主鎖帰属用実験
- 側鎖帰属用実験
- H/D交換実験
- 構造計算用実験
- その他
詳細
1. 準備
1-1. 測定試料
実験に応じて、\(\ce{^{15}N}\)ラベル試料または\(\ce{^{13}C}\)/\(\ce{^{15}N}\)ラベル試料を用いる。大腸菌発現系によるラベル蛋白質の作製については、本アーカイブのプロトコール(池上貴久, 蛋白質科学会アーカイブ, 1, e014 (2008))に詳しく記述されている。蛋白質は通常、0.2 – 1.0 mM程度の濃度、室温以上の温度、中性以下の pH で数日間安定に溶解していることが望ましい。試料は軽水試料(90%\(\ce{H2O}\)にロック用として10%\(\ce{D2O}\)を添加)と重水試料(100%\(\ce{D2O}\))の二種類を用いるが、まずは使用頻度の高い軽水試料を調製する。
バッファーは、観測可能な\(\ce{^{1}H}\)を含まない20 mM程度のリン酸緩衝液が一般的である。pHは低いほどアミド水素の交換が遅くシグナル強度が高まるが、pH 8以下であれば多くの場合問題ない。Tris、HEPES、酢酸などを用いる場合は、可能な限り重水素化試薬を使用する。ただし、軽水試料を用いた主鎖シグナルの帰属段階では、バッファーや添加物由来の信号は大きな妨げとならないため、非重水素化試薬を使用してもよい。一方、重水試料において蛋白質以外の\(\ce{^{1}H}\)を極力排除する必要がある場合には、リン酸バッファーであっても凍結乾燥後に\(\ce{D2O}\)へ再溶解し、交換性\(\ce{^{1}H}\)を\(\ce{D}\)に置換する処理を行うことがある。また、試料濃縮に頻用される限外ろ過膜の洗浄が不十分だと、グリセロールが混入するため注意する。\(\ce{NaCl}\)などの塩を50 – 200 mM程度添加することも多いが、高塩濃度は特に低温プローブで感度低下を引き起こすため、必要最小限にとどめる。さらに、試料中に常磁性金属イオンが混入すると測定結果に悪影響を及ぼすため、使用する水や器具の洗浄には気を配る必要がある。
蛋白質用NMRチューブはSHIGEMI社製のミクロセルが事実上の標準になっている(NMRのメーカーに応じて底部の長さが異なるので要注意)。液量は250 – 300 μL程度必要である。SHIGEMIチューブの外管に、途中にサンプルがなるべく付かないように試料を入れる。管底まで届く長いパスツールピペットを使うか、ピペットチップ/シリンジに外径2 mm程度のテフロンチューブを付けたもので行うと便利である。内管で溶液を上から押さえて気泡を抜く。このとき溶液を泡立てないよう注意する。内管を固定するように外管の口の部分をパラフィルム等で巻いて閉じる。
1-2. 1D測定
NMR装置の測定ソフトウエアでは、以下に示すパラメータ調整用のセットアップを保存しておき、最初にそれをロード(またはその場所に移動)してから作業を開始するのがよい。まずプローブを測定温度に設定し、軽水試料の入ったNMRチューブをスピナーの正しい位置にセットして投入する。温度は20 – 40℃程度に設定することが多いが、蛋白質が安定ならば高い方がよい。スピナーは回転させない。試料投入後数分待ってからプローブのチューニング、ロック及びシムの調整を行う。現代の装置ではこれらの調整は自動で行えるのが普通である(チューニング及びシムのマニュアル調整にはある程度習熟が必要である)。
調整後、まず\(\ce{^{1}H}\)の90°パルス幅を測定する。これは過去に測定した試料であっても一連の実験前には改めて測定する。\(\ce{^{1}H}\)の出力パワーを通常使用する最大値に設定し、レシーバーゲインを最低にし、まず1 – 2 μs程度のパルス長を使って一回積算で水のシグナルを観測し、フーリエ変換の位相を補正する。続いてパルス長を変えて測定することを繰り返し、360°パルスに相当するパルス長(信号が消失する値。180°パルスでも信号が消失するが360°の方が良い)を探す。スペクトルではなく、FIDを見て判断してもよい。\(\text{90°パルス長} = \text{360°パルス長}/4\)であり、通常10マイクロ秒前後になる。得られた値を以降の測定に用いる。高塩濃度の試料では長くなる傾向があるが、異常に長い場合は試料調製のミスや装置の劣化も考えられる。高感度プローブの場合、水のシグナルが強すぎて90°パルス測定が出来ない場合がまれにある。その際はpre-saturation法によって水信号を消去し、バッファー由来信号等で測定する。後の実験で\(\ce{^{1}H}\)のデカップリングやミキシングを使う場合は、用いる90°パルス幅が決まっているので、それを与える出力パワーを探す作業が必要な場合もある。
次に\(\ce{^{1}H}\)の1Dスペクトルを測定する。\(\ce{^{1}H}\)の観測中心を水に設定し、観測幅は15 ppm程度とする。Pre-saturation/Watergate/Excitation sculptingなどの手法により、信号を消去し測定する。水信号が消去できればレシーバーゲインを大きくでき、数回の積算で蛋白質由来のシグナルが十分観測できるはずである。消去効率は、水の線形と、照射位置(観測中心周波数)に大きく依存するので、上手く行かない場合は、シム調整と照射位置の最適化を行う。
得られた1Dスペクトルのピークの分布を確認する。0 ppm以下にメチル基由来のピークがある場合は立体構造が形成されている可能性が高い。また5 – 6 ppmにHα由来ピークがある場合はおそらくβシートが形成されている。溶媒によってはブロードな蛋白質ピークとは別にTris等バッファー成分に由来するシャープなピークが観測される。精製・濃縮過程でグリセロールが混入すると、3.6 ppm付近にピークが現れる。実際に1Dスペクトルを細かく解析することは少ないが、試料の状態を確認するためにも毎回測定しておきたい。
1-3. \(\ce{^{15}N}\) HSQC測定
次いで、二次元\(\ce{^{15}N}\) HSQCスペクトルを測定する。これは蛋白質のNMRにおいて最も基本となるスペクトルである。試料としては\(\ce{^{15}N}\)ラベルした蛋白質あるいは\(\ce{^{15}N}\)/\(\ce{^{13}C}\)ラベルした蛋白質を用いる。\(\ce{^{1}H}\)は観測中心を水に設定し、観測幅は12 ppm程度、観測時間は50 – 100 msとする(例えば600 MHzの装置では観測幅13.3 ppm = 8000 Hz、複素ポイント数512点で観測時間は64 msになる)。観測時に\(\ce{^{15}N}\)をパルスデカップリングするので、\(\ce{^{1}H}\)の観測ポイントを不用意に多くすると装置・試料が過熱するため注意する。\(\ce{^{15}N}\)は観測中心を117 ppm前後、観測幅30 ppm程度にし、複素ポイント数64 – 128程度にする。積算の間隔は1秒程度にする。通常2 – 8回の積算で測定できる。\(\ce{^{13}C}\)ラベルした蛋白質の場合はt1期に\(\ce{^{13}C}\)デカップリングが必要である。\(\ce{^{15}N}\)の観測中心、観測幅は得られたスペクトルでピークがスペクトル端にかからないよう調整する。通常、t1の初期値がパルスプログラム中で適切に設定され、その結果\(\ce{^{15}N}\)軸の位相補正(0次/1次)は\(0/0\)あるいは\(-90/180\)になり、\(\ce{^{15}N}\)の観測幅からはみ出したピークは位相が歪むこと無く折り返す。そうならない場合は、t1の初期値を正しく設定する必要がある。
1-4. \(\ce{^{15}N}\) HSQCの評価
測定後のフーリエ変換・解析は、NMR装置付随のソフトウエアを用いるか、公開されている別の解析ソフトを用いる。よく使われているフーリエ変換ソフトの一つにNIHで開発されたNMRPipeがある。NMRPipeでは、データ処理の各段階(ウインドウ関数・ゼロフィリング・フーリエ変換・位相補正・ベースライン補正など)を行うコマンドをシェルスクリプトファイルに順次記述する。データはコマンド間を受け渡されていくに従って処理され、最終的にスペクトルとなる。得られたスペクトルはNMRPipe付属のNmrDrawで表示できる。ピークの重なり部分などの見え方は、処理方法(ウインドウ関数、リニアプレディクションなど)で大きく変わることに留意する。
\(\ce{^{15}N}\) HSQC(図1に例を示す)で観測されるのはPro及びN末端以外の主鎖アミド、Trp側鎖の\(\varepsilon\ce{NH}\)、Asn/Glnの側鎖\(\delta/\varepsilon\ce{NH2}\)である。場合によってはArgの\(\varepsilon\ce{NH}\)も観測される(Lysの\(\zeta\)アミノ基、Hisのイミダゾール\(\ce{NH}\)、Argの\(\eta\ce{NH2}\)を観測するためには、特定の条件や専用の実験を要する)。Asn/Glnの側鎖\(\ce{NH2}\)は\(\ce{^{1}H}\)が6.5 – 8.0 ppm、\(\ce{^{15}N}\)が109 – 115 ppmに\(\ce{^{15}N}\)の化学シフトが等しい2つのピークとして現れる。またNHD由来(ロック用重水に起因)の小さな信号がこぶのようにピークの上に見えることがある。Trp側鎖は\(\ce{^{1}H}\)が10 ppm程度に現れる。Argの\(\varepsilon\ce{HN}\)は\(\ce{^{15}N}\)の化学シフトが設定範囲外(85 ppm付近)であるため、折り返しの形で現れる。主鎖アミドでは、Gly由来ピークがスペクトル上部(高磁場側、\(< 110\ \mathrm{ppm}\))に現れるのが特徴であり、測定範囲の設定によってはスペクトル下部に折り返しとして観測される。Ser、Thrも似た傾向を示す。また経験上、C末端アミノ酸のHNは\(\ce{^{1}H}\)が8 ppm、\(\ce{^{15}N}\)が125 – 130 ppmあたりに強く観測されることが多い。
以上を考慮し、主鎖アミド基のピークがどのように見えているか注意する。まず、残基数(-Proの数 - 1)程度のピークが見えているはずである。また、ピークどうしの重なりが少なく分散しており、シグナル強度に極端な差が無いことが望ましい。ピークの\(\ce{^{1}H}\)化学シフトが狭い(〜1 ppm、\(< 8.5\ \mathrm{ppm}\))範囲に集中している場合は、おそらく立体構造が形成されていない。蛋白質複合体の一部や、マルチドメインの一部を切り出した蛋白質では、構造多形が生じるためにシグナル強度差が大きい。これらのケースではドメイン境界の再検討も含め、更なる条件最適化が勧められる。また、比較的高濃度で測定するため、通常は問題にならない分子間の弱い非特異的会合がスペクトルの質を落とす場合があり、濃度を下げた方が結果的に良いこともある。\(\ce{^{15}N}\) HSQCが綺麗に測定できない試料はその後の測定でも不都合が生じる場合が多いので、安易に妥協せず十分検討した方が結果的に早く実験・解析を進めることができる。なお、ピークの分散が少ない天然変性状態の試料でも、主鎖アミドの帰属は可能であることが多い(吉村優一, 蛋白質科学会アーカイブ, 8, e080 (2015))。
2. 主鎖帰属用実験
主鎖帰属は\(\ce{^{13}C}\)/\(\ce{^{15}N}\)ラベル試料を用い複数の3D NMR実験を組み合わせて行う。よく用いられるHNCACB, CBCA(CO)NHの組み合わせではHN, N, Cα, Cβが帰属できる。HNCACB, CBCA(CO)NHだけでは曖昧さが解消されない場合、C’の化学シフトを使うHNCO, HN(CA)COを用いれば解決できる。また、溶解度等の問題で十分な感度が得られない場合、比較的感度の高いHNCA, HN(CO)CAを援用する場合もある。これらの3D NMR実験で最も高感度なのはHNCOであり、低感度なのはHNCACB及びHN(CA)COで、その差は一桁程度あるが、通常後者でも1 – 2日程度で測定できる。
各測定とも通常複素ポイント数として\(\ce{^{15}N}\)軸を32程度、\(\ce{^{13}C}\)軸を32 – 64程度で測定する。\(\ce{^{1}H}\)、\(\ce{^{15}N}\)の観測中心、観測幅は\(\ce{^{15}N}\) HSQCと揃える。\(\ce{^{13}C}\)の観測中心、観測幅は見ている部位(Cα, Cα/β, C’)に応じて変えるが、組み合わせるスペクトル間では揃える。設定可能なパラメータの数は多く、また同じ実験でもパルスプログラムとしては異なる実装が多数ありうるのでここでパラメータの詳細を述べることは困難である。実際には一度設定すれば変更が必要ないパラメータも多いので、初期セットを専門家に設定してもらうとよい。また、パルス長などは一つ設定すれば残りはそこから計算されるようになっている場合が多い。いずれにせよ用いるパルスプログラムに対応した個々のパラメータの意味を把握しておくことは重要である。
3D NMRの解析にはPOKY、CcpNmr、NMRViewJ、Kujiraなど蛋白質のNMR解析用ソフトが不可欠であり、詳しい操作はそれぞれのマニュアル等を参照されたい。基本的には\(\ce{^{15}N}\) HSQCで得られるHN, Nの化学シフトの組が各残基のIDとして機能すると考える。このIDを実際のアミノ酸番後に結びつけるのが目標である。実際は\(\ce{^{15}N}\) HSQCで完全に重なっているピークもいくつか存在するので、高感度で分離の良い3D HNCO法を援用するとよい。すなわちHNCOでピークをピックした後、その位置を\(\ce{^{15}N}\) HSQC上に投影すれば、HNCOではC’で分離しているが、HSQCでは重なっているピークの存在を明らかにできる。そのようにして、まずHN, Nの化学シフトの組をリストアップする。
各HN, Nの化学シフト位置で3Dスペクトルを2次元の短冊状に切り出した断片をstripと呼び、これを並べて解析する。HNCACBではstripの縦軸が\(\ce{^{13}C}\)軸となり、HN(i), N(i)に対してCα(i), Cβ(i), Cα(i–1), Cβ(i–1)にピークが得られる。このときCβ由来ピークの位相は反転しており(表示ではCαと異なる色で示される)。一方、CBCA(CO)NHではCα(i–1), Cβ(i–1)のピークが得られ、CαとCβの位相は同じである。
この2種類のスペクトルを対応づけながら隣接残基に由来するstripを探し、連続するstripを同定・並べ替えることで帰属を伸張していく。Cα/Cβの化学シフト値はアミノ酸の判別に有効であり、連結されたセグメントが配列上のどこに対応するかを決定できる。解析ソフトウエアには、このようなスペクトルの同時解析を半自動的に行う機能が備わっている。化学シフトの重複等により、明確な帰属が困難である場合、アミノ酸選択的標識(あるいは非標識)試料の調製も考慮に値する。これによって、HSQCピークが特定のアミノ酸(群)に由来することが判明すると、帰属の曖昧さの解消に役立つ。
順当に作業を進めれば、HSQCで観測される主鎖アミドピークのほぼすべてを帰属できる。さらに、主鎖帰属が完了した段階で、Chemical Shift Indexを用いて二次構造を推定することが可能となる。またNMRPipeに付属するプログラムTALOS+によって、主鎖の二面角を推定することも可能である。
3. 側鎖帰属用実験
側鎖の帰属は、3D実験を用いて主鎖の帰属を側鎖へと伸張することで行う。まずHBHA(CBCACO)NHにより、主鎖アミドの帰属に基づいてHαとHβを同定する。残りのアルキル部分については、H(CCO)NHおよびC(CCO)NHを用いると簡便である。これらで十分なピークが得られない場合や、Pro前残基のようにアミドと連結できない箇所では、CCH-TOCSY、HCCH-TOCSY、CCH-COSY、HCCH-COSYを利用し、Hα/CαやHβ/Cβとの交差ピークから帰属を行う。分子量が比較的小さい(\(< 15\ \mathrm{k}\))蛋白質では、3D \(\ce{^{15}N}\)-separated TOCSYにより側鎖Hをある程度帰属することもできる。
すべての側鎖ピークを一意に帰属できるとは限らない。特にメチレンのプロキラルHはこの段階で立体特異的に区別できないが、後のNOE解析や構造計算過程で帰属できる場合がある。一方、メチル基はシャープで強いピークを与えるため、Ala、Thr、Ile、Leu、Valの帰属は比較的容易である。メチル基由来NOEは構造計算において有用な制約となるため、優先的に帰属するのが望ましい(Metのεメチル基も明瞭に観測できるが、通常の測定では他の残基内シグナルとの連結は困難である)。全体を俯瞰するにはconstant-time \(\ce{^{13}C}\) HSQCが有効である。\(\ce{^{15}N}\) HSQCに比べピークの重なりは大きいが、α位およびメチル基は分離が比較的良く、帰属結果の確認に役立つ。なお、アミドを利用しない実験(CCH-TOCSY, HCCH-TOCSY, CCH-COSY, HCCH-COSY)は、\(\ce{D2O}\)試料を用いた方が高品質なスペクトルが得られる。ただし軽水試料との間で化学シフトに微妙な差が生じるため、両方の条件で\(\ce{^{13}C}\) HSQCを測定し比較するのが望ましい。
芳香環側鎖については、まずδ位あるいはε位とβ位とを(HB)CB(CGCD)HD、(HB)CB(CGCDCE)HEによって連結し、その後芳香環用CCH-TOCSYにより環内の相関をたどる。ただし芳香環の化学シフトは重なりが大きく、一意に帰属できないことも多い。Asn/Glnの側鎖\(\ce{NH2}\)基については、HNCACBを改変したパルスシーケンスにより、残基内メチレン部位との連結を確認できる。以上の手順で困難な部位は、NOEの解析過程で補助的に帰属する。
4. H/D交換実験
本実験は構造決定に必須ではないが、他に得られないユニークな情報が得られるので解説する。まず\(\ce{^{15}N}\)ラベルした試料を準備する。基本的には帰属等に用いる試料と同等なものでよい。H/D交換の過程は\(\ce{^{15}N}\) HSQCスペクトルで追跡するため、\(\ce{^{15}N}\) HSQCが数分〜一時間程度で測定可能な濃度の試料が必要である。
[手法1] 凍結乾燥
エッペンドルフチューブ等に入れた試料を液体窒素などで瞬間的に凍結させ、真空デシケータ等の中で吸引して乾燥させる。これを凍結前と同体積の重水に溶解すれば、凍結前と同じ組成の溶液となる(バッファーが非揮発性の場合)。コツは、性能の良い真空設備を使い、容器を傾けるなどして凍結時の表面積を稼いで蒸発速度を上げることである。
[手法2] 脱塩カラム
重水で調製したNMR測定用バッファーを用意する。ゲル濾過を利用したNAP-5(Cytiva)等の脱塩カラムを重水バッファーで平衡化しておき、NMR試料を通すことで溶媒を置換する。試料が希釈されるので、必要ならばAmicon Ultra(メルク)などで濃縮する。作業中のH/D交換を遅くするため、なるべく低温で実施する。
[手法3] 希釈・再濃縮
重水バッファーで数倍に希釈し、Amicon Ultraなどで元の体積まで濃縮することを3、4回繰り返す。[手法2]同様、低温で実施する。膜へ吸着する試料や沈殿し易い試料には向かない。
各手法とも一長一短がある。[手法1]の凍結乾燥後試料は保存可能で重水を添加すれば直ぐ測定できる。しかし設備が必要であり、試料によっては変性して再溶解できない場合がある。従って、H/D交換が速く、変性しにくい試料に向いている。[手法2]、[手法3]は作業中にもH/D交換が起こってしまうので、交換が極めて速い部位の測定は難しいが、蛋白質中の二次構造領域は十分交換が遅く実用上はそれほど問題にならない。失敗が少なく、設備も必要でないため無難な方法である。置換後はすぐにHSQCスペクトルを測定する。手法にもよるが、交換開始後15分から2時間程度の時点で測定することになる。ここで交換されず残っている部位は、水素結合によって保護されていると示唆される。その後必要に応じてHSQCを測定しH/D交換の様子を追跡する。速いH/D交換速度を定量的に求めたい場合など、実験中に交換が起こると望ましくないことがある。その場合は、試料のpHを下げてH/D交換をクエンチする。一般に主鎖アミドの易動性プロトンのH/D交換はpH 2.5前後で最も遅く(〜\(0.1 \mathrm{min}^{-1}\))、pHがそこから1変化すると交換速度は1桁増えて中性では\(1 \times 10^{3} \mathrm{min}^{-1}\)程度に達する。
5. 構造計算用実験
構造計算に最も重要な情報は、近接(\(< 5\ –\ 6\ \text{Å}\))の\(\ce{^{1}H}\)間距離を与えるNOEである。通常は3D \(\ce{^{15}N}\)-separated NOESYおよび3D \(\ce{^{13}C}\)-separated NOESYにより測定する。\(\ce{^{15}N}\)-separated NOESYでは任意の\(\ce{^{1}H}\)からアミドプロトンへのNOEを観測し、\(\ce{^{15}N}\)または\(\ce{^{13}C}\)/\(\ce{^{15}N}\)ラベル試料を用いるが、前者の方が高品質なスペクトルが得られる。\(\ce{^{13}C}\)-separated NOESYでは任意の\(\ce{^{1}H}\)から\(\ce{^{13}C}\)結合プロトンへのNOEを観測する。原則として\(\ce{^{13}C}\)/\(\ce{^{15}N}\)ラベル試料を用い、重水試料で測定する方がスペクトル品質は高い。側鎖\(\ce{^{13}C}\)の化学シフトや1JCH値はアルキル基と芳香環で大きく異なるため、測定条件を分けて取得する。NOESYの重要なパラメータであるmixing timeは、通常50 – 100 msに設定する。
各スペクトルからは数千以上のNOEピークが得られるため、解析には専用ソフトウエアの利用が不可欠である。まずピークをピックし、既存の化学シフトテーブルやスペクトル内での対称性を用いて、NOEを\(\ce{^{1}H}\)ペアに帰属する。化学シフト値のみからすべてのNOEを一意に帰属することは困難であるため、信頼度の高いNOEを優先的に利用してラフな構造を推定し、その結果をもとに残りのNOEの帰属を絞り込む、という反復作業を行う。相同蛋白質の既知構造や、AlphaFold等による予測構造も参考になるだろう。解析ソフトはNOEに基づく\(\ce{^{1}H}\)間距離制限を出力し、これをCYANA、Xplor-NIH、CNSなどの構造計算ソフトに入力する。これらのソフトでは、帰属が曖昧なNOEも複数候補の距離制限として利用可能であり、距離の\(-6\)乗平均を用いることで、構造計算中に矛盾の少ない拘束が優先される仕組みとなっている。構造計算と並行して、二面角制限(化学シフトやJ値から推定)やH/D交換実験で得られた水素結合情報を導入することもできる。
NOEは定量性に乏しいため、通常はstrong/medium/weak(必要に応じvery weak)に分類し、それぞれ2.5/3.5/5.0(/6.0)Å程度の距離上限として扱う。計算結果では入力制限が必ずしも満たされず、違反(violation)が生じる場合がある。その際は制限をより緩いカテゴリに移す、あるいは削除して再計算するが、無批判に修正するのは危険である。多くの場合、違反は化学シフト帰属の誤りに由来するため、スペクトルの再確認が必要である。
また、距離制限情報の特性上、入力条件を満たす構造解は複数存在する。そのため、通常はランダムに生成した数十個の初期構造から計算を行い、結果を重ね合わせて評価する。構造計算とNOE帰属を繰り返すことで、主鎖原子座標のRMSDが0.5 Å程度に収束する構造集合が得られる。得られた構造はラマチャンドランプロット等で妥当性を評価する。重ね合わせた平均構造をAmberなどでエネルギー最適化し、代表構造とすることもある。
6. その他
J値による二面角の推定
3JHN-Haを求めてφ角の値を推定することができる。実験としてはHMQC-J、HNHA法などがある。HNHAはHαの帰属にも役立つ。また、3JHa-Hb、3JN-Hb、3JC’-Hbからχ1角を推定するとともに、プロキラルなβプロトンを立体特異的に帰属することができる。そのための実験としてはHNHB、HN(CO)HB法などがある。これらの測定は感度が低く、必ずしも全ての残基が観測できるわけではない。
Val/Leuメチル基の立体特異的帰属
大腸菌発現系を用いて蛋白質をラベルしている場合は、大腸菌のアミノ酸代謝系の性質を利用することで簡便に帰属できる。すなわち、炭素源であるglucoseの一部(10 – 15%)を\(\ce{[^{13}C]}\)-glucoseにして発現させ、得られた試料の\(\ce{^{13}C}\)-HSQCを測定するだけでよい。すると、\(\ce{^{13}C}\)γ1(Val)あるいは\(\ce{^{13}C}\)δ1(Leu)は付け根の\(\ce{^{13}C}\)と同じglucose分子から生じているため、カップリングが観測される。
ダイナミクスの評価
緩和時間測定によって分子内部のダイナミクスを評価することは、蛋白質のNMR解析において重要なテーマであり多くの研究が行われている。簡便な評価法としては、\(\ce{\{^{1}H\}}\)-\(\ce{^{15}N}\) NOE測定がある。これは\(\ce{^{1}H}\)共鳴を飽和させて\(\ce{^{1}H}\)-\(\ce{^{15}N}\)間の定常状態NOEを測定するもので、ps – nsオーダーの速い分子内運動が存在する部位を明らかにできる。そのような部位では\(\ce{^{1}H}\)間のNOEも観測されにくく、立体構造も収束しないのが普通である。また、何点か温度を変えて\(\ce{^{15}N}\)-HSQCを測定してみるのもよいだろう。構造多形がある場合、温度変化によって構造間での平衡が変化しスペクトルの変化となって現れる。より詳しい多形解析には、緩和分散法(谷中冴子ら, 蛋白質科学会アーカイブ, 7, e076 (2014))が有効である。
残余双極子カップリング
バイセル、ファージ粒子などの非等方的な物質を溶媒中に混在させるか、引き延ばしたアクリルアミドゲル中に蛋白質を浸透させると、蛋白質がそれらとの相互作用により溶液中で弱く配向する。すると、通常溶液中では消失している双極子カップリングが僅かに復活して測定可能になる。このカップリング値は双極子間ベクトルと分子の配向軸との角度に依存するため、NOEやJ等から得られる局所的・相対的な構造情報とは異なる性格の立体構造情報として有力である。測定自体は容易だが、蛋白質によっては適切な配向溶媒系を見つけるのが難しい。金属結合部位を持つ、あるいは金属結合タグを人為的に導入した蛋白質では、磁気的な異方性が大きいランタニドイオンを結合させることによっても蛋白質を配向させることができる。
常磁性シフト・緩和
常磁性の金属イオンやTEMPOなどのラジカルを、結合タグの導入や化学修飾という形で付加することにより、それらの常磁性シフト・緩和効果を構造情報として用いることが可能になる。これらは最大数十Åの影響範囲を持つため、NOEでは得られない遠位の構造情報を与える。
工夫とコツ
試料調製
100 – 200 mM程度の硫酸塩がNMRスペクトルを顕著に改善する場合がある。おそらく蛋白質の構造形成を促し多形を抑制している。ただし、低温プローブでは感度が低下する。他に物性を改善しうる添加物として、スルフォベタイン類、中性アミノ酸(Gly等)、あるいはArg/Gluの等モル混合物などがある。これら両性イオンはプローブの感度に悪影響を与えないが、Gly以外はD化体が無いか高価である。アミノ酸のような補償溶質が共存すると、耐熱性や長期安定性が向上することを経験している。他に、必要に応じてEDTA、DTT、TCEPなどを添加してもよい。ミセルサイズの小さい界面活性剤(CHAPSなど)を添加する場合もある。グリセロールなど溶液の粘度が増加する添加物は避ける。
以上を考慮し、まず少量の蛋白質を用いて溶液条件を探索する。結晶化プレートや透析ボタンを用いて、沈殿が生成しない条件をスクリーニングすることや、ゲル濾過、超遠心、光散乱などの方法で分散状態を評価するのも有効である。ただしNMRでは、他の手法では観測できない弱い分子間相互作用や、僅かに存在する準安定コンフォメーションの影響で線形が悪化している場合もあり、結局NMRで条件検討する方が早いこともありえる。また、測定条件を最適化して、本来見たい条件から離れてしまっては本末転倒という面もある。試料は、見かけ上溶解していても最後に0.22 μmのスピンフィルターを通しておく。NMRチューブは蛋白質の吸着を防ぐためシリコナイズ処理しておくと良い。
同じような分子量の蛋白質でも、スペクトルの質は蛋白質によって様々である。また、ハードウエアの状態やソフトウエアの設定によってスペクトルの質は劇的に変化することがある。可能であればユビキチンなど質の良いスペクトルを与えることが分かっている試料をポジティブコントロールとして用意しておくことが勧められる。ラベルユビキチンは市販されている。
装置定数
以下は定期的に確認することを推奨する。
化学シフト基準値:蛋白質試料には標準物質を加えず、別に外部標準試料を作って測定しておく。標準物質としては2,2-dimethylsilapentane-5-sulfonic acid(DSS)の水溶液を用いる。DSSの共鳴周波数を測定し、\(\ce{^{1}H}\)の0 ppmとする。\(\ce{^{13}C}\)、\(\ce{^{15}N}\)に関しては\(\ce{^{1}H}\)の周波数の25.144953%、10.1329118%をそれぞれ0 ppmとする。
\(\ce{^{13}C}\)、\(\ce{^{15}N}\)のパルス長:それぞれ、分光計メーカー提供の標準サンプルで測定する。自作する場合は\(\ce{[^{15}N]}\)-尿素と\(\ce{[^{13}C]}\)-メタノールをd6-DMSOに溶かして調製する。異なるパワーでの90度パルス長を測り線形性を確かめる。
温度:NMRプローブの温度は、分光計メーカー提供の標準サンプルで校正しておく。温度制御系はサンプル内部の温度ではなく、サンプル周囲のエアーの温度を測っている。従ってプローブの種類や外部エアー流量・温度によって表示値と実際の温度とのずれは変化する。
測定
主鎖帰属以降の過程では必ず複数の実験を組み合わせることになる。可能なら同じ試料で連続して実験を行う方がよい。少なくともHNCACBとCBCA(CO)NHなどペアで解析する実験は連属して行うべきである。その際、安定性に劣る試料の場合は多次元測定の合間に\(\ce{^{15}N}\) HSQCを測定し、試料の状態をチェックできるようにしておくとよい。複数の3D実験を連続して行う場合、実際の測定前に必ずそれぞれの実験の1Dもしくは2Dバージョンを測定し、異常が無いか確認するとともに、実験ごとにレシーバーゲインを適切に設定しておく。長期間測定していると試料内で気泡が生じてシムが合わなくなり、水信号が消えなくなってレシーバーがオーバーフローすることがあるので注意する。
濃度が高く測定感度に問題がない試料では、非線形サンプリングにより、測定時間を大幅に短縮できることがある。また、また、試料の縦緩和を最適化し、積算繰り返し時間を最小化するパルスシーケンス(SOFAST, BEST系)も時間短縮に有効である。一方でこれらの手法には、設定や処理が複雑化する、特有のアーティファクトや定量性の低下、装置の設定に敏感になる、といった面もある。通常の測定・解析に習熟後、適切に活用するのがよいと思われる。
フーリエ変換
ピークの見え方は、フーリエ変換(FT)時のパラメータで大きく変化する。変換後の分解能とS/N比は相反するため、それぞれを優先した条件で変換したスペクトルを用意し、両方を見ながら解析するのも良いだろう。
NMR装置付随のソフトを使わない場合、FT時のfirst point補正係数を正しく設定する必要があるが、間違え易いので注意点を述べる。対象軸の観測幅をswとすると、展開時間の初期値は、通常は0、0.5/sw(half dwell)あるいは1/sw(full dwell)から有限パルス長に起因する補正値を引いた値に設定されている。どの設定になるかは実験の種類や観測幅及びパルスシーケンスの実装などに依存する。それに合わせて、FT時のfirst point補正係数(cとする)、位相補正p0/p1を以下のように設定する。
- 0の場合
c = 0.5, p0 = 0, p1 = 0 - half dwellの場合
c = 1.0, p0 = -90, p1 = 180
full dwellの場合は、fidを右に1点シフト(1点目が2点目になる。NMRpipeならnmrPipe -fn ZF -pad 1 | nmrPipe -fn RS -rs 1 -sw)し、1点目はbackwardリニアプレディクションで構築(nmrPipe -fn LP -before -pred 1)した後、0の場合同様に c = 0.5、p0 = 0、p1 = 0 でFTする。
Webサイト
ケミカルシフトデータベース
- BMRB: https://bmrb.io/
ソフトウエア
- NMRPipe: https://www.ibbr.umd.edu/nmrpipe/
- POKY: https://sites.google.com/view/pokynmr
- NMRViewJ: https://nmrfx.org/
- CYANA: https://www.cyana.org/
- xplor-NIH: https://nmr.cit.nih.gov/xplor-nih/
- CNS: https://cns-online.org/
文献
- Cavanagh, J. et al., Protein NMR Spectroscopy, Elsevier Academic Press, Burlington, San Diego and London (2007)
- T.D.W.クラリッジ, 有機化学のための高分解能NMRテクニック(竹内敬人・西川実希 訳), 講談社(2004)
-
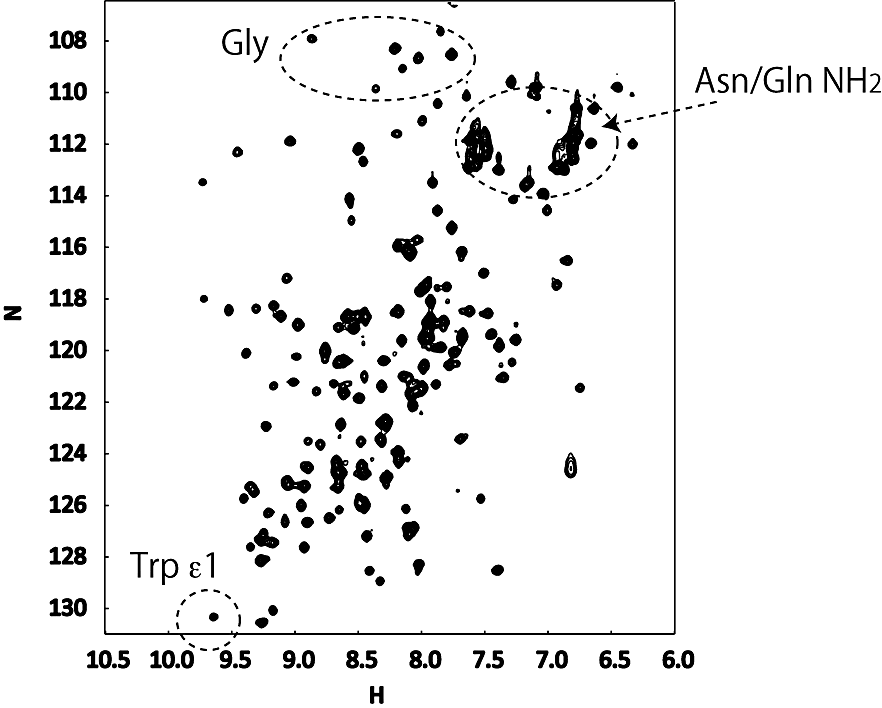
図1:蛋白質の\(\ce{^{15}N}\)-HSQCスペクトルの例
