

概要
緩和分散法はミリ秒からサブミリ秒の化学交換(構造変化)を解析できるNMR法である。この時間領域には、蛋白質―リガンド相互作用や酵素反応などの生体反応が含まれるため蛋白質の機能を調べる上で大変有効な測定法である。蛋白質の構造変化という言葉でイメージされるのは多くの場合、結晶構造で観測されるような基質結合前後の開閉構造、ドメイン間の大きな移動だろう。そして、構造変化の時間領域や溶液中での構造の存在比という概念は見落とされる事が多い。しかしながら、溶液中には結晶構造では観測されない低存在比の中間構造、ドメイン内の局所的な構造変化が存在し、様々な時間領域で動的構造変化が起きている。そして、近年は溶液中での動的構造はタンパク質の機能発現に関わるという考え方が定着してきている(1, 2)。溶液中での動的構造変化を原子レベルで解き明かす上で、緩和分散法は頼りとなる数少ない手法である。そこで、本プロトコールでは測定が一番容易な15N標識に話を絞り、測定手法を簡単に紹介したい。緩和分散法の原理は参考文献(3-5)に詳細に解説されているため、本稿では実践的な測定試料調製、NMR測定、データ解析、結果の解釈について解説する。
装置・器具・試薬
- 静磁場の異なる2つのNMR(2核測定が可能)
- 5ΦNMRチューブ
- 15N標識(必要に応じて2H標識)タンパク質
実験手順
1)測定試料調製
2)NMR測定
3)データ解析
4)結果の解釈
実験の詳細
1)測定試料調製
まず、15N標識試料の調製を行う。15N標識試料の測定により、蛋白質主鎖の構造が動的に変化する部位を特定できる。調製方法は蛋白質科学会アーカイブ(池上貴久、蛋白質科学会アーカイブ, 1、e014 (2008))を参照されたい。緩和分散法では高感度のデータが求められるため、高濃度で安定なサンプルが必要となる。0.5 mM程度で少なくとも4日間は安定なサンプルが望ましい。なお、帰属用には別途、13C、15N標識試料の調製、測定、及び解析(吉田卓也、蛋白質科学会アーカイブ, 3、e059 (2010))が必要であるが、緩和分散が先に確認できると、帰属など、測定と解析に1~2ヶ月を要する後の実験に対するモチベーションが高くなるため、まずは15N標識サンプルを調製して緩和分散を測定することを勧める。サンプルは精製後、測定バッファーに濃縮器を用いた限外ろ過法にて透析する。測定はバッファー条件にも敏感なため、ロック用の5%程度のD2Oを含むNMRバッファーを多めに調製しておき、毎回それを使用することが推奨される。また、緩和分散の温度依存性を調べる場合、温度に依存してpHが変化するバッファーは使用できないため、リン酸バッファーやHEPESバッファーなどをお勧めする。15N緩和分散実験を行う上で、重水素化は考慮にいれるべき安定同位体標識のオプションである。特に、アミノ酸数が150以上ある場合は、シグナルの緩和が速くなり解析が困難になるため、重水素化が望まれる。重水素化すると高分子量のタンパク質の測定に有効なTROSYバージョンの緩和分散法(6)が使用可能になる。
2)NMR測定
NMR測定の基本的な操作は蛋白質科学会アーカイブ(吉田卓也、蛋白質科学会アーカイブ, 3、e059 (2010))を参照されたい。緩和分散法は、特に温度に敏感であるため、液体窒素や液体ヘリウムは実験前夜までに充填して、翌朝から測定を開始するようにする。測定の前には必ずメタノール(またはエチレングリコール)を用い温度校正を行う。次に実サンプルに変えて測定のセットアップを進める。まず、パルスプログラムは、cw CPMG(7)、または重水素化していればTROSYバージョン(6)を使用する。まず、シムの最適化を行い、続いてパルスの校正を行う。15Nの180°パルスが正確でないと人工的な緩和分散が得られてしまう場合があるため15Nの180°パルスは実サンプルに対して決める必要がある。パルス校正は一次元HSQCを用いる。パルスの校正方法は1Hの180°パルスと同様である。
次に、観測される水シグナルが最小になるようにグラジエント強度とwater-flip-back pulseのパラメータを最適化する。水消しが悪い場合、感度良く測定できない。また、スペクトル上に水シグナルが大きく現れるとベースラインが歪む。強いベースライン補正(4次関数など)を適用すれば、歪みは改善されるが、微妙な強度変化を定量化する緩和分散では、シグナル強度まで変えてしまう強いベースライン補正は避けたほうが良い。Bruker BioSpinの場合はgsと言うインタラクティブにNMRパラメータを最適化するコマンドがあり、gsを用いてwater-flip-backパルスプログラムのshaped pulseパワーを最適化する。これにより、水が非常に良く消えてダイナミックレンジを稼げ、S/Nが良くなるため緩和分散の質が向上する。
最後に、サンプルやNMRに凝集や分解等に伴う経時変化があるとあたかも緩和分散があるかのように見える場合があるため、測定順番をシャッフルする。また、それぞれの測定の前には、磁場の均一性を最適化するコマンドを挟む。これによって、それぞれの2次元NMR測定はほぼ同じ磁場の均一性から開始されることになり、磁場の経時変化由来の誤差を小さくすることができる。一つの磁場における測定のみでは求めるパラメータに対してデータが少なく、精度が悪いため、2つの磁場を用いて測定を行う。これにより、正確なパラメータを得ることができる。
3)データ解析
まず、NMRスペクトルをフーリエ変換する。スペクトルの基本的な処理方法については蛋白質科学会アーカイブ(吉田卓也、蛋白質科学会アーカイブ, 3、e059 (2010))を参照していただきたい。linear predictionはシグナル強度を変化させるため使用してはならない。また、定量性を保ちつつ測定を短縮する方法としては、非線形サンプリングとSIFT法と呼ばれるNMRスペクトルのプロセス法を組み合わせる方法が成功を収めている(8)。この手法は、測定時間を1/3以下に短縮できるため、不安定で経時変化のあるサンプルの緩和分散測定に有効である。続いてシグナル強度を定量化する。シグナルの位置とシグナル強度を算出するソフトには様々ある。当研究室で独自に開発したGLOVEと呼ばれるプログラムに内蔵されたpkfitというプログラムでシグナルの位置とシグナル強度を算出している。ピークトップを中心とした3x3ポイントのボックスを設定し、3x3の各グリッドポイントにおけるシグナルの高さの和を強度としている。次にフィッティングを行う。幾つかのフッティングプログラムがウェブから入手可能であるが、当研究室ではGLOVEを使用している(9)。GLOVEでは、フィッティングにおける極小値問題を解決するためにMonte Carlo minimizationを採用しており、従来用いられているグリッドサーチ法よりも非常に早く最適解にたどり着く(図1)。
4)結果の解釈
緩和分散の測定、解析によってダイレクトに得られるバラメータは構造変化の大きさを表す化学シフト差Δω、低存在比構造の割合pb、観測される構造変化の速度kexである。ここから、順方向、逆方向の構造変化速度を算出できる。これらの値から、溶液中で起きる動的構造変化について、定量的に議論することが可能となる。筆者らは緩和分散法を用いて、ヒト主要組織適合遺伝子複合体(HLA)の解析を行なった。結晶構造では強固な構造を取っていることが予測されるような領域で構造変化が起きていることを明らかにした(図2)。そして、構造変化速度の解析から、観測した構造変化は、HLAが安定な構造を取るのに必要な動きであることが明らかとなった。緩和分散法の測定では、より早い/遅いタイムスケールの動きを観測することができないという制限もあるが、他のNMR測定法を駆使することで様々な時間領域の構造変化を観測することも可能である。様々な時間領域の構造変化の解析を通して、蛋白質の動きが機能に与える影響が明らかとなっていくであろう。
工夫とコツ
サンプル数について
緩和分散は2つの磁場で測定を行う必要がある。タンパク質の経時変化を最小にするために、可能であれば全く同じNMRサンプルを2つ用意し、2つの磁場で同時に測定を開始する。
サンプル管の準備について
測定中にNMRチューブ内に泡が発生すると緩和分散カーブが汚くなるため、NMRチューブに詰めたサンプルは、とくにシゲミチューブの場合、しっかりと脱気する。脱気をするには、まずはNMRチューブに試料を入れ、シゲミチューブの内管を挿入する。この時点でも往々にして泡があるため、まずは、しばらく低温で静置して泡がなくなるのを待ち、その後、ゴムパッキンやパラフィルムなどで内管を固定せずに、デシケーターに入れ、真空ラインで引く。泡が見えてきたら、デシケーターから出し、内管を上下して泡を潰す。再度真空引きし、泡が出なくなるまでこの作業を繰り返す。
文献
- Bhabha, G. et al., Science, 8, 234-8 (2011)
- Sugase, K., et al., Nature, 447, 1021-5 (2007).
- 菅瀬 謙治, 蛋白質核酸酵素, 52, 945-51 (2007)
- 菅瀬 謙治, 生化学, 80, 754-58 (2008)
- 菅瀬 謙治, 生物物理, 48, 279-81 (2008)
- Vallurupalli, P. et al., Proc. Natl. Acad. Sci. USA. 104, 18473-7 (2007)
- Hansen, D. F. et al., J. Phys. Chem. B, 112, 5898-904 (2008)
- Matsuki, Y. et al., J. Phys. Chem. B, 115, 13740-5 (2011)
- Sugase, K. et al., J. Biomol. NMR, 56, 275-83 (2013)
-
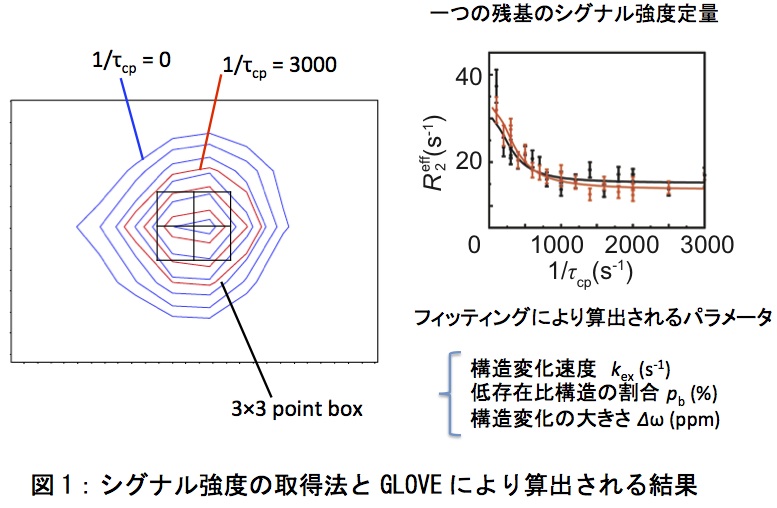
図1 -

図2
概要
緩和分散法はミリ秒からサブミリ秒の化学交換(構造変化)を解析できるNMR法である。この時間領域には、蛋白質―リガンド相互作用や酵素反応などの生体反応が含まれるため蛋白質の機能を調べる上で大変有効な測定法である。蛋白質の構造変化という言葉でイメージされるのは多くの場合、結晶構造で観測されるような基質結合前後の開閉構造、ドメイン間の大きな移動だろう。そして、構造変化の時間領域や溶液中での構造の存在比という概念は見落とされる事が多い。しかしながら、溶液中には結晶構造では観測されない低存在比の中間構造、ドメイン内の局所的な構造変化が存在し、様々な時間領域で動的構造変化が起きている。そして、近年は溶液中での動的構造はタンパク質の機能発現に関わるという考え方が定着してきている(1, 2)。溶液中での動的構造変化を原子レベルで解き明かす上で、緩和分散法は頼りとなる数少ない手法である。そこで、本プロトコールでは測定が一番容易な15N標識に話を絞り、測定手法を簡単に紹介したい。緩和分散法の原理は参考文献(3-5)に詳細に解説されているため、本稿では実践的な測定試料調製、NMR測定、データ解析、結果の解釈について解説する。
装置・器具・試薬
- 静磁場の異なる2つのNMR(2核測定が可能)
- 5ΦNMRチューブ
- 15N標識(必要に応じて2H標識)タンパク質
実験手順
1)測定試料調製
2)NMR測定
3)データ解析
4)結果の解釈
実験の詳細
1)測定試料調製
まず、15N標識試料の調製を行う。15N標識試料の測定により、蛋白質主鎖の構造が動的に変化する部位を特定できる。調製方法は蛋白質科学会アーカイブ(池上貴久、蛋白質科学会アーカイブ, 1、e014 (2008))を参照されたい。緩和分散法では高感度のデータが求められるため、高濃度で安定なサンプルが必要となる。0.5 mM程度で少なくとも4日間は安定なサンプルが望ましい。なお、帰属用には別途、13C、15N標識試料の調製、測定、及び解析(吉田卓也、蛋白質科学会アーカイブ, 3、e059 (2010))が必要であるが、緩和分散が先に確認できると、帰属など、測定と解析に1~2ヶ月を要する後の実験に対するモチベーションが高くなるため、まずは15N標識サンプルを調製して緩和分散を測定することを勧める。サンプルは精製後、測定バッファーに濃縮器を用いた限外ろ過法にて透析する。測定はバッファー条件にも敏感なため、ロック用の5%程度のD2Oを含むNMRバッファーを多めに調製しておき、毎回それを使用することが推奨される。また、緩和分散の温度依存性を調べる場合、温度に依存してpHが変化するバッファーは使用できないため、リン酸バッファーやHEPESバッファーなどをお勧めする。15N緩和分散実験を行う上で、重水素化は考慮にいれるべき安定同位体標識のオプションである。特に、アミノ酸数が150以上ある場合は、シグナルの緩和が速くなり解析が困難になるため、重水素化が望まれる。重水素化すると高分子量のタンパク質の測定に有効なTROSYバージョンの緩和分散法(6)が使用可能になる。
2)NMR測定
NMR測定の基本的な操作は蛋白質科学会アーカイブ(吉田卓也、蛋白質科学会アーカイブ, 3、e059 (2010))を参照されたい。緩和分散法は、特に温度に敏感であるため、液体窒素や液体ヘリウムは実験前夜までに充填して、翌朝から測定を開始するようにする。測定の前には必ずメタノール(またはエチレングリコール)を用い温度校正を行う。次に実サンプルに変えて測定のセットアップを進める。まず、パルスプログラムは、cw CPMG(7)、または重水素化していればTROSYバージョン(6)を使用する。まず、シムの最適化を行い、続いてパルスの校正を行う。15Nの180°パルスが正確でないと人工的な緩和分散が得られてしまう場合があるため15Nの180°パルスは実サンプルに対して決める必要がある。パルス校正は一次元HSQCを用いる。パルスの校正方法は1Hの180°パルスと同様である。
次に、観測される水シグナルが最小になるようにグラジエント強度とwater-flip-back pulseのパラメータを最適化する。水消しが悪い場合、感度良く測定できない。また、スペクトル上に水シグナルが大きく現れるとベースラインが歪む。強いベースライン補正(4次関数など)を適用すれば、歪みは改善されるが、微妙な強度変化を定量化する緩和分散では、シグナル強度まで変えてしまう強いベースライン補正は避けたほうが良い。Bruker BioSpinの場合はgsと言うインタラクティブにNMRパラメータを最適化するコマンドがあり、gsを用いてwater-flip-backパルスプログラムのshaped pulseパワーを最適化する。これにより、水が非常に良く消えてダイナミックレンジを稼げ、S/Nが良くなるため緩和分散の質が向上する。
最後に、サンプルやNMRに凝集や分解等に伴う経時変化があるとあたかも緩和分散があるかのように見える場合があるため、測定順番をシャッフルする。また、それぞれの測定の前には、磁場の均一性を最適化するコマンドを挟む。これによって、それぞれの2次元NMR測定はほぼ同じ磁場の均一性から開始されることになり、磁場の経時変化由来の誤差を小さくすることができる。一つの磁場における測定のみでは求めるパラメータに対してデータが少なく、精度が悪いため、2つの磁場を用いて測定を行う。これにより、正確なパラメータを得ることができる。
3)データ解析
まず、NMRスペクトルをフーリエ変換する。スペクトルの基本的な処理方法については蛋白質科学会アーカイブ(吉田卓也、蛋白質科学会アーカイブ, 3、e059 (2010))を参照していただきたい。linear predictionはシグナル強度を変化させるため使用してはならない。また、定量性を保ちつつ測定を短縮する方法としては、非線形サンプリングとSIFT法と呼ばれるNMRスペクトルのプロセス法を組み合わせる方法が成功を収めている(8)。この手法は、測定時間を1/3以下に短縮できるため、不安定で経時変化のあるサンプルの緩和分散測定に有効である。続いてシグナル強度を定量化する。シグナルの位置とシグナル強度を算出するソフトには様々ある。当研究室で独自に開発したGLOVEと呼ばれるプログラムに内蔵されたpkfitというプログラムでシグナルの位置とシグナル強度を算出している。ピークトップを中心とした3x3ポイントのボックスを設定し、3x3の各グリッドポイントにおけるシグナルの高さの和を強度としている。次にフィッティングを行う。幾つかのフッティングプログラムがウェブから入手可能であるが、当研究室ではGLOVEを使用している(9)。GLOVEでは、フィッティングにおける極小値問題を解決するためにMonte Carlo minimizationを採用しており、従来用いられているグリッドサーチ法よりも非常に早く最適解にたどり着く(図1)。
4)結果の解釈
緩和分散の測定、解析によってダイレクトに得られるバラメータは構造変化の大きさを表す化学シフト差Δω、低存在比構造の割合pb、観測される構造変化の速度kexである。ここから、順方向、逆方向の構造変化速度を算出できる。これらの値から、溶液中で起きる動的構造変化について、定量的に議論することが可能となる。筆者らは緩和分散法を用いて、ヒト主要組織適合遺伝子複合体(HLA)の解析を行なった。結晶構造では強固な構造を取っていることが予測されるような領域で構造変化が起きていることを明らかにした(図2)。そして、構造変化速度の解析から、観測した構造変化は、HLAが安定な構造を取るのに必要な動きであることが明らかとなった。緩和分散法の測定では、より早い/遅いタイムスケールの動きを観測することができないという制限もあるが、他のNMR測定法を駆使することで様々な時間領域の構造変化を観測することも可能である。様々な時間領域の構造変化の解析を通して、蛋白質の動きが機能に与える影響が明らかとなっていくであろう。
工夫とコツ
サンプル数について
緩和分散は2つの磁場で測定を行う必要がある。タンパク質の経時変化を最小にするために、可能であれば全く同じNMRサンプルを2つ用意し、2つの磁場で同時に測定を開始する。
サンプル管の準備について
測定中にNMRチューブ内に泡が発生すると緩和分散カーブが汚くなるため、NMRチューブに詰めたサンプルは、とくにシゲミチューブの場合、しっかりと脱気する。脱気をするには、まずはNMRチューブに試料を入れ、シゲミチューブの内管を挿入する。この時点でも往々にして泡があるため、まずは、しばらく低温で静置して泡がなくなるのを待ち、その後、ゴムパッキンやパラフィルムなどで内管を固定せずに、デシケーターに入れ、真空ラインで引く。泡が見えてきたら、デシケーターから出し、内管を上下して泡を潰す。再度真空引きし、泡が出なくなるまでこの作業を繰り返す。
文献
- Bhabha, G. et al., Science, 8, 234-8 (2011)
- Sugase, K., et al., Nature, 447, 1021-5 (2007).
- 菅瀬 謙治, 蛋白質核酸酵素, 52, 945-51 (2007)
- 菅瀬 謙治, 生化学, 80, 754-58 (2008)
- 菅瀬 謙治, 生物物理, 48, 279-81 (2008)
- Vallurupalli, P. et al., Proc. Natl. Acad. Sci. USA. 104, 18473-7 (2007)
- Hansen, D. F. et al., J. Phys. Chem. B, 112, 5898-904 (2008)
- Matsuki, Y. et al., J. Phys. Chem. B, 115, 13740-5 (2011)
- Sugase, K. et al., J. Biomol. NMR, 56, 275-83 (2013)
