

概要
近年、天然変性蛋白質が生体内の重要なイベント(転写や翻訳の制御、膜融合、細胞周期制御など)に関与していることが明らかとなってきた(1)。溶液NMRは蛋白質の構造、物性、機能を原子レベルで明らかにするための重要な分光法のひとつである。しかしながら、特定の立体構造をもつ蛋白質と比べて変性した蛋白質は測定核周辺の環境の磁気的な多様性に乏しいため、NMR信号の分散が小さく(図1A、B)信号帰属に苦労することが多い。本稿では、NMR信号分散の小さい変性蛋白質について、4次元以上のNMR実験に頼ることなく容易かつ明確に主鎖信号帰属をおこなう手法を紹介する。
イントロダクション
蛋白質の主鎖NMR信号の連鎖帰属については蛋白質科学会アーカイブ(吉田卓也、蛋白質科学会アーカイブ, 3, e059 (2010))に詳しく記述されている。折り畳まれた蛋白質では側鎖の13C核は化学的環境に応じて化学シフトの分散が大きいため、3次元(3D)NMR測定(たとえば、HNCACBおよびCBCA(CO)NH)により得られる13Cαおよび13Cβ核の残基内(i)・残基間(i-1)の相関(図1C)に基づいて化学シフトのマッチングから主鎖信号の連鎖帰属をおこなう(図1D)。しかしながら、変性蛋白質は特定の立体構造をとらないため、図1Eに示すように、これらの化学シフト値は自身のアミノ酸の種類にのみ依存する。特に、繰り返し配列が含まれると13Cαおよび13Cβ核の化学シフト値のみからユニーク(一意的)に帰属をおこなうのは極めて困難である。
一方、変性蛋白質の主鎖カルボニル炭素(13C’)およびアミド窒素(15NH)の化学シフトは自身および隣接するアミノ酸の種類によって影響を受けるため、13Cαおよび13Cβと比較して化学シフト値の分散は大きい(図2A)。また、これらの核は変性蛋白質では横緩和時間が長いため、化学シフト展開時間を長く設定することにより、分解能のよいNMRスペクトルを取得することができる。以下で紹介するNMRパルス系列(3D (HN)CO(CO)NH,(H)N(COCO)NH)を用いることで、最大7残基の相関を得ることができる。したがって、NMR信号分散の小さい変性蛋白質について、化学シフトの縮重が問題になることなく、曖昧さのないユニークなマッチングが可能となる。すなわち、容易かつ明確に変性蛋白質の主鎖信号帰属をおこなうことができる。
装置・器具・試薬
- 1H/13C/15N三重共鳴プローブを装着したNMR
- 安定同位体標識(13C/15N)した蛋白質
実験手順
1)測定試料の調製
2)NMR測定
2-1)2D 1H-15N HSQC、H(N)CO
2-2)3D HNCO
2-3)3D (HN)CO(CO)NH、(H)N(COCO)NH
2-4)3D HNCACB、(H)CC(CO)NH
3)NMR信号帰属
実験の詳細
1)測定試料の調製
本稿で紹介する信号帰属法では、均一13C/15N二重標識した蛋白質試料の調製が必要である。大腸菌による同位体標識蛋白質の発現については蛋白質科学会アーカイブ(池上貴久、蛋白質科学会アーカイブ, 1, e014 (2008))に記述されている。特定の立体構造をもつ蛋白質と比較して、変性蛋白質は局所の回転拡散運動が速いため、横緩和時間が比較的長い。そのため、0.2mM程度の蛋白質濃度でも十分に感度のよい3D NMRスペクトルを取得することができる。一方、アミド水素は溶媒中の水素との交換から保護されていないために、生理的条件下(たとえばpH7.4、37℃)ではアミド水素の信号を検出できないことが多い。そのため、pHを低くする、温度を下げる、などの検討が必要である。ただし、極低温プローブを装着した高感度測定が可能な装置があればアミド水素の代わりに13C直接観測が可能であるため、以下で紹介するパルス系列を1H観測から13C観測に変更することにより、生理的条件でのNMR測定も可能である。
筆者は、蛋白質試料に50μM程度の4,4-dimethyl-4-silapentane-1-sulfonate(DSS)を内部標準物質として添加している。理由は、DSSのメチル水素の1H共鳴周波数を0ppmとすることと、信号の線形から磁場の均一性がシムコイルによって最適化されているか否かを確認するためである。ただし、蛋白質が正電荷を帯びている場合は、DSSとの静電相互作用のため、磁場の均一性がよくてもDSS信号の線幅が広幅化する場合がある。
2)NMR測定
ここでは、パルス幅やパワー校正などについては省略する。以下では、主鎖アミド(1HN、15NH)をベースにしたNMR信号帰属のための2Dおよび3D NMRスペクトル測定について述べる。
2-1)2D 1H-15N HSQC、H(N)CO
NMR信号帰属において2D 1H-15N HSQCは基本となるスペクトルである。そのため、特に15N軸はできるだけ分解能よく測定する。HSQCスペクトルから、後の3D NMR実験で必要な15N軸の観測幅と中心周波数を決める。筆者が11.7テスラ(1Hラーモア周波数500MHz)NMRを用いて P. fluorescens 由来の変性蛋白質FapCのNMR信号帰属をおこなった際は、15N観測幅23ppm(1.17kHz)、複素ポイント数256(取り込み時間:約220ms)でHSQC測定を行っている。また、15N化学シフト展開期間に13C核をデカップルする必要がある。1H核の観測については、1HN軸の線幅は1Hα核との3J結合によって広幅化されるため、観測時間は120ms程度で十分である。これ以上観測時間を長くする場合は観測期間中の15Nデカップリングによる温度上昇に気を付ける。
次に、カルボニル炭素(13C’)を化学シフト標識する測定をおこなう。最初に、感度の最もよいHNCOスペクトルを測定するが、まずは2D版H(N)COを測定し、後の3D NMR実験で必要な13C軸の観測幅と中心周波数を決める。1H-15NHSQCスペクトルと同様、13C軸はできるだけ分解能よく測定する。13C’化学シフト展開期間は1Hβ核などとのJ結合により信号が広幅化するため、1Hをデカップルすることで高分解能スペクトルを得ることができる(図2B)。FapCのNMR信号帰属では、13C観測幅6ppm(0.75kHz)、複素ポイント数256(取り込み時間:約340ms)でH(N)CO測定を行った。
2-2)3D HNCO
上で決定した15NHおよび13C’の中心周波数および観測幅で、高分解能の3D HNCOを測定する。15N核に関して、constant timeにより化学シフト展開時間が限られている場合、高分解能スペクトルを取得することができない。このような場合、化学シフト展開をsemi-constant time(2,3)に変更することで化学シフト展開時間を延長できるため、高分解能3D HNCOスペクトルの取得が可能となる(図2B)。また、13C’化学シフト展開期間は1H核をデカップルする。
2-3)3D (HN)CO(CO)NH、(H)N(COCO)NH
13C’の残基内(i)・残基間(i-1)の相関はHN(CA)COおよびHNCO測定から得られるが、変性蛋白質では信号の分散が小さく、多くの化学シフトが縮重している(図3A)。この問題に対して、以下で紹介する3D (HN)CO(CO)NH実験によって3残基以上の化学シフト相関を得ることにより、図3Bに示すように縮重が解消される。
3D (HN)CO(CO)NHは、主鎖カルボニル間の 3JC’C’ 結合を介した磁化移動により、残基間の相関を得る実験である(図4A)。このパルス系列はGrzesiek and Bax(4)により論文発表されている。13C’混合期間中の緩和による信号強度の減少が抑制された同種核混合パルス系列(MOCCA-XY16、図4B)を用いることで、長期間(最大500ms程度)のミキシングによって最大で7残基の相関を得ることができる(図4C)ので、化学シフト値の縮重が問題になることなく帰属できる。
高分解能で(HN)CO(CO)NHスペクトルを得るためのパルス系列(Brukerコード)は、http://www.protein-nmr.org に掲載されている。オリジナルのパルス系列からの変更点は、(I)13Cおよび15N核の化学シフト展開をconstant timeからsemi-constant timeに変更、(II)13C化学シフト展開期間中の1Hデカップリングの導入、(III)同種核混合パルス系列をDIPSI3からMOCCA-XY16に変更、などである。詳細は、文献5に記述されている。
変性蛋白質では15NH核についても横緩和時間が比較的長く、13Cαや13Cβ核と比べると化学シフト値の分散がよい。したがって、15NH化学シフト値も信号帰属に利用できる。(H)N(COCO)NH実験の磁化移動は、13C’のかわりに15NH化学シフト標識する以外は、上で述べた(HN)CO(CO)NHと同じである(図4A)。(H)N(COCO)NHのBrukerパルス系列についても http://www.protein-nmr.org に掲載している。
2-4)3D (H)CC(CO)NH、HNCACB
変性蛋白質では13Cαおよび13Cβ核の化学シフトは自身のアミノ酸の種類のみに依存し、同じアミノ酸では化学シフトの縮重のため、残基内・残基間の相関からユニークにNMR信号を帰属するのは困難である。その一方で、側鎖13C核の化学シフトは隣接するアミノ酸の種類による影響が小さいため、アミノ酸残基のランダムコイルの化学シフト値との比較からアミノ酸の種類の同定が可能である。その際、CBCA(CO)NHのかわりに(H)CC(CO)NH (3)を測定することによって、13Cαや13Cβだけではなくすべての脂肪族13C核由来の信号を得ることができるので、より確実にアミノ酸の種類を同定することができる。また、図1Eに示すように、HNCACBスペクトルから、アミノ酸残基の連結パターン(たとえばVal-Alaなど)も明らかにすることができる。これらのパルス系列においても、15N核の化学シフト展開時間がconstant timeによって限られているために高分解能スペクトルを取得することができない場合、化学シフト展開をsemi-constant timeに変更して化学シフト展開時間を延長することにより、高分解能スペクトルを得ることができる。
3)NMR信号帰属
データをフーリエ変換してNMRスペクトルを得る。変性蛋白質の場合、図1Bのように1H-15N HSQCスペクトル上では多くのピークが重なっている。そのため、基本となる2Dスペクトルとして、1H-15N HSQCのほかに3D HNCOの2D投影スペクトルを用いる。特に、図2Aで示すようにHNCOスペクトルの2D 13C-15N投影図は2D 1H-15N HSQC(図1B)と比べてピーク分散がかなりよい。フーリエ変換する際、スペクトルの信号強度の定量性は重要ではないので、スペクトルの分解能をあげるために、筆者はすべての間接観測軸について線形予測(linear prediction、LP)を用いている(ただし、信号強度の定量的な解析が必要な場合は、LPは使用しない)。
変性蛋白質の信号帰属の一例を図5に示す。測定条件下(pH2.0、37℃)において、図5Aの1H-15N HSQC上で丸で囲ったピークは、2つの信号が重なっている。これらの信号について、HNCACB(図5B)および(H)CC(CO)NHスペクトルのストリッププロットからアミノ酸配列がAla-Alaであることがわかった。しかしながら、FapCには8つのAla-Ala配列が含まれている。これらの13Cαおよび13Cβ核の化学シフトはすべて縮重しているため、信号を帰属することができない。(HN)CO(CO)NHおよび(H)N(COCO)NHスペクトルを測定することにより、連続する5残基の13C’および15N化学シフトの相関を得ることができ、曖昧さなくユニークに帰属することができる(図5C)。
工夫とコツ
静磁場強度
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルを高分解能で得るためには、磁場強度は大きい方がよいように思われるが、高磁場では13C’の化学シフト異方性相互作用による緩和が問題となる。200残基程度以下の蛋白質であれば、1Hラーモア周波数500~700MHz(B0=11.7~17.6テスラ)程度のNMRで、十分にピーク分散のよいスペクトルを得ることができる。
パルスキャリブレーション
筆者は通常1Hパルスのみを校正している。13Cおよび15Nパルスについては、標準試料で校正した180度パルス幅が実試料の180度パルス幅と大きく異なるかを確認する程度である。確認は1D版のHSQCを用いている。実試料と標準試料でパルス幅やパワーが大きく異なる場合は、実試料で校正した値を用いる。
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルの13C’混合時間
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルから得られる相関は、混合期間の緩和速度や3JC’C’結合の大きさに依存する。そのため、どの程度の混合時間に設定すればよいかは、試料や磁場強度によって異なる。筆者が500MHzNMRで実験をおこなう際は、混合期間を300~500ms程度に設定し、2D版スペクトル((HN)CO(CO)(N)H、(H)N(COCO)(N)H)を測定して対角ピーク、交差ピークが十分な信号強度で得られているかを確認する。高磁場では交差ピークの信号強度が小さくて検出が困難な場合があるので、その際は混合期間を短縮して、再度2Dスペクトルを確認する。
一部のアミノ酸配列領域で横緩和時間が短い等の理由により13C’混合による磁化移動効率が悪く交差ピークが検出されない場合は、13C’混合期間を短縮したスペクトルを測定するのもよいが、次に述べるようにHN(CA)COスペクトルやHN(CA)NNHスペクトルからも残基間の相関を得ることができる。
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルで交差ピークが検出できない
局所的にポリペプチド鎖がヘリックスを形成すると、(HN)CO(CO)NHスペクトルや(H)N(COCO)NHスペクトルにおいて交差ピークが検出されない場合がある。その理由として、(I)ヘリックスを形成することにより回転拡散運動が遅くなり、横緩和速度が上昇する、(II)カルボニル炭素間の3JC’C’結合はポリペプチド鎖の二面角(φ)に依存し、ヘリックスでは3JC’C’値が小さい、ことが挙げられる。このような場合には、3D HN(CA)COやHN(CA)NNHスペクトルを得ることによって13C’および15NH化学シフトの残基間の相関を得ることができる。これらの3Dスペクトルにおいても、15N核の化学シフト展開をsemi-constant timeにすることで、高分解能のスペクトルを得ることができる。
アスパラギン側鎖との相関
(H)N(COCO)NHスペクトルで、どの主鎖15NH核の化学シフト値とも一致しない信号強度の小さな交差ピークが112ppm付近にあらわれることがある。このピークは、13C’混合期間中の側鎖カルボニル炭素から主鎖への磁化移動により得られるピークである。ただし、これらのピークは側鎖アミド基の2つの水素のうち一方が重水素に置換されている場合(15NHD)にのみ検出される。すなわち、信号強度は小さく、磁場のロックのために添加する重水の濃度に依存する。また、このときの15N核の化学シフト値は重水素置換により同位体シフトしているので、1H-15NHSQCスペクトル上の側鎖信号と照らし合わせるときは注意が必要である。
試料の温度上昇
(HN)CO(CO)NHや(H)N(COCO)NH実験では、長期間の混合パルス系列やデカップリングパルスにより、測定試料の温度が上昇する場合がある。熱電対でモニターしている温度は試料内の温度と一致していないことがあるので注意が必要である。筆者が500msの混合時間での実験において、スキャン毎の水とDSSの1H化学シフト差から試料の温度上昇を見積もったところ、ダミースキャンの間に熱電対の示す温度が目標温度で一定になっていても、試料内の温度は0.5℃高いことがわかった。試料温度を厳密に調整する必要がある場合には、あらかじめ測定による温度上昇を調べ、測定時の目標温度を温度上昇する分だけ下げておく等の工夫が必要である。
蛋白質の重水素化
変性蛋白質では13C’や15NHの横緩和時間が長いため、HNCO、(HN)CO(CO)NH、(H)N(COCO)NH実験において、間接観測軸の化学シフト展開時間を長くすることにより、高分解能のスペクトルを得ることができる。その一方、直接観測する1HNの線幅は1HNと1Hαの3J結合のため、13C’や15Nの線幅と比べて広い。この問題に対して、α水素を重水素化した蛋白質を調製し、1H信号観測時間を長くすることで1HNの線幅をシャープにすることができる。このとき、15Nデカップリングによる温度上昇に注意する必要があるが、変性蛋白質では15NH核の化学シフト分散が小さいため、比較的弱いパワーでも効率よくデカップリングできる。別の方法として、信号取得中のバンド選択的同種核(BASH)デカップリング(6)によって分解能を向上することも可能であると思われる。
非線形サンプリング
高分解能3D NMRスペクトル測定では、測定に長時間を要する。試料の経時変化が問題となる場合は、非線形サンプリングをおこなうことで測定時間を短縮できる。
文献
- Wright, P.E. and Dyson, H.J., J. Mol. Biol., 293, 321-31 (1999)
- Grzesiek, S. and Bax, A., J. Biomol. NMR, 3, 185-204 (1993)
- Logan, T.M., et al., J. Biomol. NMR, 3, 225-231 (1993)
- Grzesiek, S. and Bax, A., J. Biomol. NMR, 9, 207-11 (1997)
- Yoshimura, Y., Kulminskaya, N.V., Mulder, F.M.M., J. Biomol. NMR, 61, 109-21 (2015)
- Ying, J., Roche, J., Bax, A., J. Magn. Reson., 241, 97-102 (2014)
謝辞
ここで紹介したプロトコールは、Frans A. A. Mulder准教授(オーフス大学)の指導の下で確立した方法である。本プロトコールの作成にあたり、安定同位体標識蛋白質(ユビキチン、α-シヌクレイン、FapC)を、それぞれ、Tania A. Nielsen様、Camilla B. Andersen様、Brian S. Vad博士(いずれもオーフス大学)から提供していただいた。また、EMBO Long-Term Fellowship(ALTF 687-2013)の留学支援に感謝申し上げる。
-
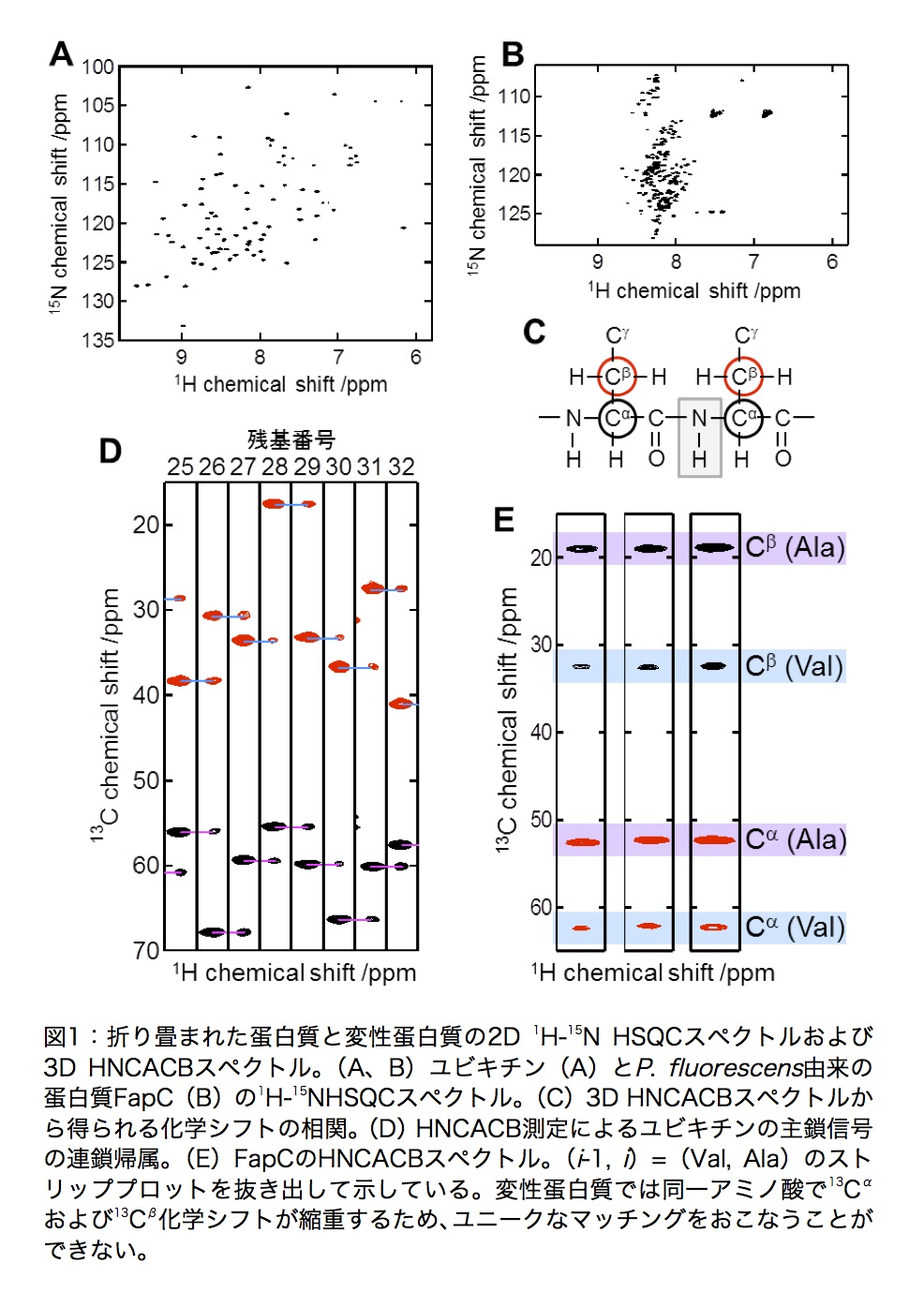
図1 -
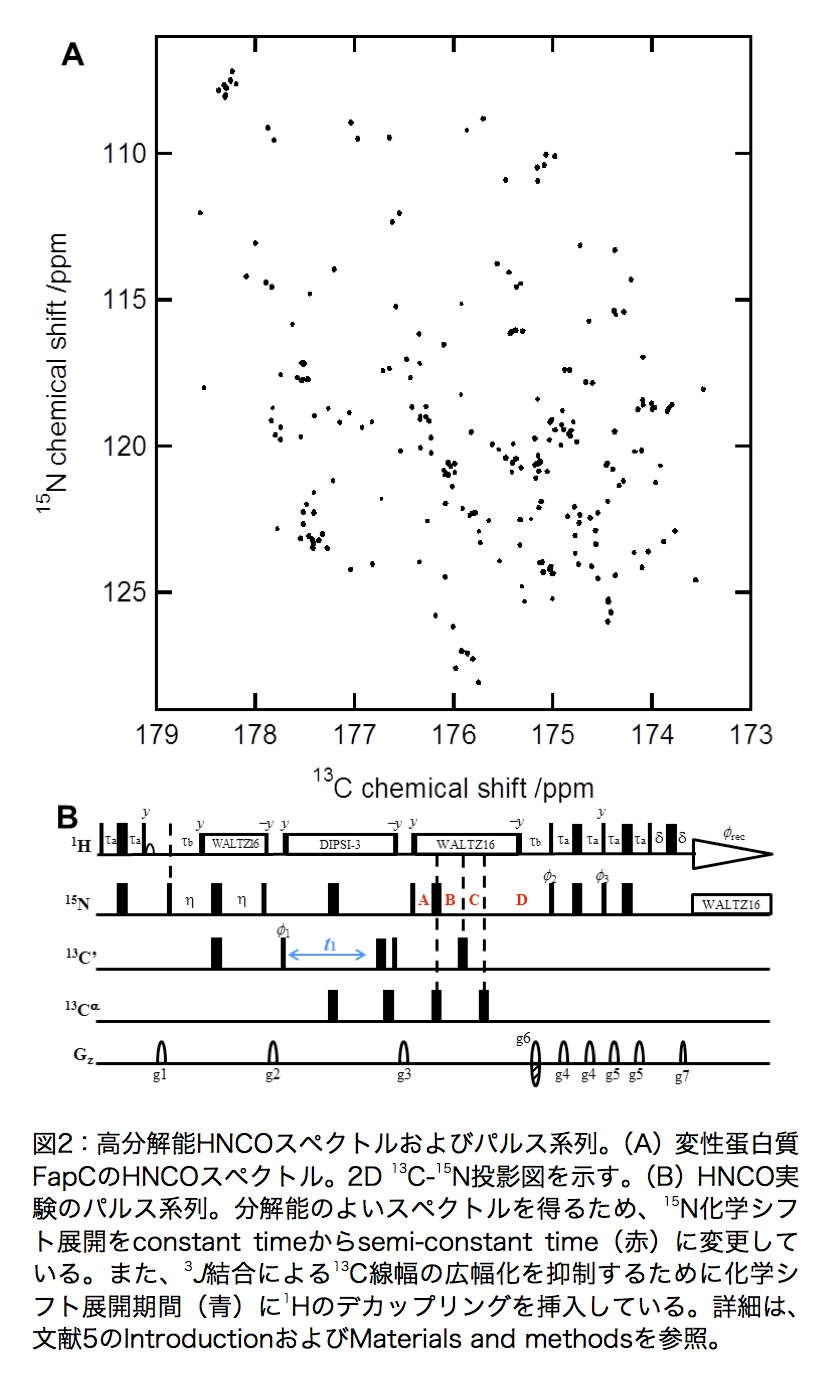
図2 -

図3 -
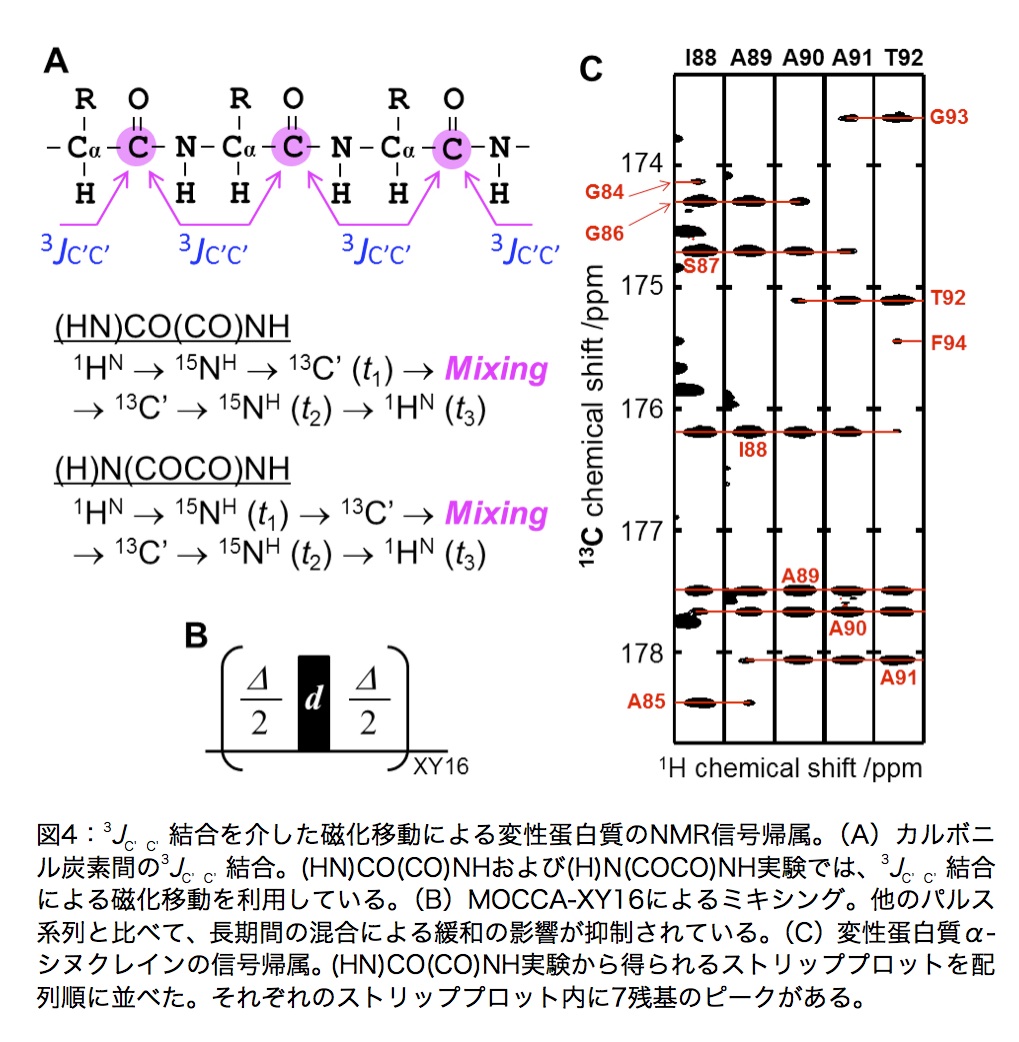
図4 -
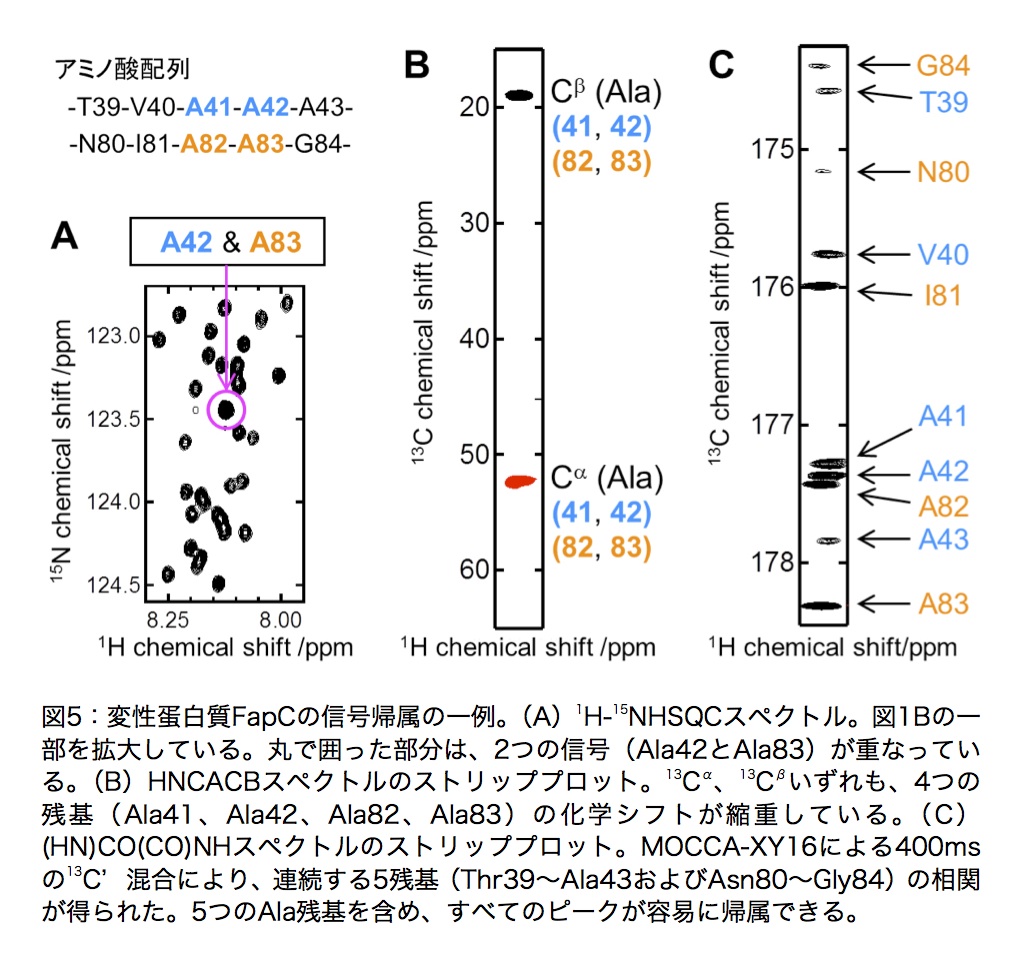
図5
概要
近年、天然変性蛋白質が生体内の重要なイベント(転写や翻訳の制御、膜融合、細胞周期制御など)に関与していることが明らかとなってきた(1)。溶液NMRは蛋白質の構造、物性、機能を原子レベルで明らかにするための重要な分光法のひとつである。しかしながら、特定の立体構造をもつ蛋白質と比べて変性した蛋白質は測定核周辺の環境の磁気的な多様性に乏しいため、NMR信号の分散が小さく(図1A、B)信号帰属に苦労することが多い。本稿では、NMR信号分散の小さい変性蛋白質について、4次元以上のNMR実験に頼ることなく容易かつ明確に主鎖信号帰属をおこなう手法を紹介する。
イントロダクション
蛋白質の主鎖NMR信号の連鎖帰属については蛋白質科学会アーカイブ(吉田卓也、蛋白質科学会アーカイブ, 3, e059 (2010))に詳しく記述されている。折り畳まれた蛋白質では側鎖の13C核は化学的環境に応じて化学シフトの分散が大きいため、3次元(3D)NMR測定(たとえば、HNCACBおよびCBCA(CO)NH)により得られる13Cαおよび13Cβ核の残基内(i)・残基間(i-1)の相関(図1C)に基づいて化学シフトのマッチングから主鎖信号の連鎖帰属をおこなう(図1D)。しかしながら、変性蛋白質は特定の立体構造をとらないため、図1Eに示すように、これらの化学シフト値は自身のアミノ酸の種類にのみ依存する。特に、繰り返し配列が含まれると13Cαおよび13Cβ核の化学シフト値のみからユニーク(一意的)に帰属をおこなうのは極めて困難である。
一方、変性蛋白質の主鎖カルボニル炭素(13C’)およびアミド窒素(15NH)の化学シフトは自身および隣接するアミノ酸の種類によって影響を受けるため、13Cαおよび13Cβと比較して化学シフト値の分散は大きい(図2A)。また、これらの核は変性蛋白質では横緩和時間が長いため、化学シフト展開時間を長く設定することにより、分解能のよいNMRスペクトルを取得することができる。以下で紹介するNMRパルス系列(3D (HN)CO(CO)NH,(H)N(COCO)NH)を用いることで、最大7残基の相関を得ることができる。したがって、NMR信号分散の小さい変性蛋白質について、化学シフトの縮重が問題になることなく、曖昧さのないユニークなマッチングが可能となる。すなわち、容易かつ明確に変性蛋白質の主鎖信号帰属をおこなうことができる。
装置・器具・試薬
- 1H/13C/15N三重共鳴プローブを装着したNMR
- 安定同位体標識(13C/15N)した蛋白質
実験手順
1)測定試料の調製
2)NMR測定
2-1)2D 1H-15N HSQC、H(N)CO
2-2)3D HNCO
2-3)3D (HN)CO(CO)NH、(H)N(COCO)NH
2-4)3D HNCACB、(H)CC(CO)NH
3)NMR信号帰属
実験の詳細
1)測定試料の調製
本稿で紹介する信号帰属法では、均一13C/15N二重標識した蛋白質試料の調製が必要である。大腸菌による同位体標識蛋白質の発現については蛋白質科学会アーカイブ(池上貴久、蛋白質科学会アーカイブ, 1, e014 (2008))に記述されている。特定の立体構造をもつ蛋白質と比較して、変性蛋白質は局所の回転拡散運動が速いため、横緩和時間が比較的長い。そのため、0.2mM程度の蛋白質濃度でも十分に感度のよい3D NMRスペクトルを取得することができる。一方、アミド水素は溶媒中の水素との交換から保護されていないために、生理的条件下(たとえばpH7.4、37℃)ではアミド水素の信号を検出できないことが多い。そのため、pHを低くする、温度を下げる、などの検討が必要である。ただし、極低温プローブを装着した高感度測定が可能な装置があればアミド水素の代わりに13C直接観測が可能であるため、以下で紹介するパルス系列を1H観測から13C観測に変更することにより、生理的条件でのNMR測定も可能である。
筆者は、蛋白質試料に50μM程度の4,4-dimethyl-4-silapentane-1-sulfonate(DSS)を内部標準物質として添加している。理由は、DSSのメチル水素の1H共鳴周波数を0ppmとすることと、信号の線形から磁場の均一性がシムコイルによって最適化されているか否かを確認するためである。ただし、蛋白質が正電荷を帯びている場合は、DSSとの静電相互作用のため、磁場の均一性がよくてもDSS信号の線幅が広幅化する場合がある。
2)NMR測定
ここでは、パルス幅やパワー校正などについては省略する。以下では、主鎖アミド(1HN、15NH)をベースにしたNMR信号帰属のための2Dおよび3D NMRスペクトル測定について述べる。
2-1)2D 1H-15N HSQC、H(N)CO
NMR信号帰属において2D 1H-15N HSQCは基本となるスペクトルである。そのため、特に15N軸はできるだけ分解能よく測定する。HSQCスペクトルから、後の3D NMR実験で必要な15N軸の観測幅と中心周波数を決める。筆者が11.7テスラ(1Hラーモア周波数500MHz)NMRを用いて P. fluorescens 由来の変性蛋白質FapCのNMR信号帰属をおこなった際は、15N観測幅23ppm(1.17kHz)、複素ポイント数256(取り込み時間:約220ms)でHSQC測定を行っている。また、15N化学シフト展開期間に13C核をデカップルする必要がある。1H核の観測については、1HN軸の線幅は1Hα核との3J結合によって広幅化されるため、観測時間は120ms程度で十分である。これ以上観測時間を長くする場合は観測期間中の15Nデカップリングによる温度上昇に気を付ける。
次に、カルボニル炭素(13C’)を化学シフト標識する測定をおこなう。最初に、感度の最もよいHNCOスペクトルを測定するが、まずは2D版H(N)COを測定し、後の3D NMR実験で必要な13C軸の観測幅と中心周波数を決める。1H-15NHSQCスペクトルと同様、13C軸はできるだけ分解能よく測定する。13C’化学シフト展開期間は1Hβ核などとのJ結合により信号が広幅化するため、1Hをデカップルすることで高分解能スペクトルを得ることができる(図2B)。FapCのNMR信号帰属では、13C観測幅6ppm(0.75kHz)、複素ポイント数256(取り込み時間:約340ms)でH(N)CO測定を行った。
2-2)3D HNCO
上で決定した15NHおよび13C’の中心周波数および観測幅で、高分解能の3D HNCOを測定する。15N核に関して、constant timeにより化学シフト展開時間が限られている場合、高分解能スペクトルを取得することができない。このような場合、化学シフト展開をsemi-constant time(2,3)に変更することで化学シフト展開時間を延長できるため、高分解能3D HNCOスペクトルの取得が可能となる(図2B)。また、13C’化学シフト展開期間は1H核をデカップルする。
2-3)3D (HN)CO(CO)NH、(H)N(COCO)NH
13C’の残基内(i)・残基間(i-1)の相関はHN(CA)COおよびHNCO測定から得られるが、変性蛋白質では信号の分散が小さく、多くの化学シフトが縮重している(図3A)。この問題に対して、以下で紹介する3D (HN)CO(CO)NH実験によって3残基以上の化学シフト相関を得ることにより、図3Bに示すように縮重が解消される。
3D (HN)CO(CO)NHは、主鎖カルボニル間の 3JC’C’ 結合を介した磁化移動により、残基間の相関を得る実験である(図4A)。このパルス系列はGrzesiek and Bax(4)により論文発表されている。13C’混合期間中の緩和による信号強度の減少が抑制された同種核混合パルス系列(MOCCA-XY16、図4B)を用いることで、長期間(最大500ms程度)のミキシングによって最大で7残基の相関を得ることができる(図4C)ので、化学シフト値の縮重が問題になることなく帰属できる。
高分解能で(HN)CO(CO)NHスペクトルを得るためのパルス系列(Brukerコード)は、http://www.protein-nmr.org に掲載されている。オリジナルのパルス系列からの変更点は、(I)13Cおよび15N核の化学シフト展開をconstant timeからsemi-constant timeに変更、(II)13C化学シフト展開期間中の1Hデカップリングの導入、(III)同種核混合パルス系列をDIPSI3からMOCCA-XY16に変更、などである。詳細は、文献5に記述されている。
変性蛋白質では15NH核についても横緩和時間が比較的長く、13Cαや13Cβ核と比べると化学シフト値の分散がよい。したがって、15NH化学シフト値も信号帰属に利用できる。(H)N(COCO)NH実験の磁化移動は、13C’のかわりに15NH化学シフト標識する以外は、上で述べた(HN)CO(CO)NHと同じである(図4A)。(H)N(COCO)NHのBrukerパルス系列についても http://www.protein-nmr.org に掲載している。
2-4)3D (H)CC(CO)NH、HNCACB
変性蛋白質では13Cαおよび13Cβ核の化学シフトは自身のアミノ酸の種類のみに依存し、同じアミノ酸では化学シフトの縮重のため、残基内・残基間の相関からユニークにNMR信号を帰属するのは困難である。その一方で、側鎖13C核の化学シフトは隣接するアミノ酸の種類による影響が小さいため、アミノ酸残基のランダムコイルの化学シフト値との比較からアミノ酸の種類の同定が可能である。その際、CBCA(CO)NHのかわりに(H)CC(CO)NH (3)を測定することによって、13Cαや13Cβだけではなくすべての脂肪族13C核由来の信号を得ることができるので、より確実にアミノ酸の種類を同定することができる。また、図1Eに示すように、HNCACBスペクトルから、アミノ酸残基の連結パターン(たとえばVal-Alaなど)も明らかにすることができる。これらのパルス系列においても、15N核の化学シフト展開時間がconstant timeによって限られているために高分解能スペクトルを取得することができない場合、化学シフト展開をsemi-constant timeに変更して化学シフト展開時間を延長することにより、高分解能スペクトルを得ることができる。
3)NMR信号帰属
データをフーリエ変換してNMRスペクトルを得る。変性蛋白質の場合、図1Bのように1H-15N HSQCスペクトル上では多くのピークが重なっている。そのため、基本となる2Dスペクトルとして、1H-15N HSQCのほかに3D HNCOの2D投影スペクトルを用いる。特に、図2Aで示すようにHNCOスペクトルの2D 13C-15N投影図は2D 1H-15N HSQC(図1B)と比べてピーク分散がかなりよい。フーリエ変換する際、スペクトルの信号強度の定量性は重要ではないので、スペクトルの分解能をあげるために、筆者はすべての間接観測軸について線形予測(linear prediction、LP)を用いている(ただし、信号強度の定量的な解析が必要な場合は、LPは使用しない)。
変性蛋白質の信号帰属の一例を図5に示す。測定条件下(pH2.0、37℃)において、図5Aの1H-15N HSQC上で丸で囲ったピークは、2つの信号が重なっている。これらの信号について、HNCACB(図5B)および(H)CC(CO)NHスペクトルのストリッププロットからアミノ酸配列がAla-Alaであることがわかった。しかしながら、FapCには8つのAla-Ala配列が含まれている。これらの13Cαおよび13Cβ核の化学シフトはすべて縮重しているため、信号を帰属することができない。(HN)CO(CO)NHおよび(H)N(COCO)NHスペクトルを測定することにより、連続する5残基の13C’および15N化学シフトの相関を得ることができ、曖昧さなくユニークに帰属することができる(図5C)。
工夫とコツ
静磁場強度
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルを高分解能で得るためには、磁場強度は大きい方がよいように思われるが、高磁場では13C’の化学シフト異方性相互作用による緩和が問題となる。200残基程度以下の蛋白質であれば、1Hラーモア周波数500~700MHz(B0=11.7~17.6テスラ)程度のNMRで、十分にピーク分散のよいスペクトルを得ることができる。
パルスキャリブレーション
筆者は通常1Hパルスのみを校正している。13Cおよび15Nパルスについては、標準試料で校正した180度パルス幅が実試料の180度パルス幅と大きく異なるかを確認する程度である。確認は1D版のHSQCを用いている。実試料と標準試料でパルス幅やパワーが大きく異なる場合は、実試料で校正した値を用いる。
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルの13C’混合時間
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルから得られる相関は、混合期間の緩和速度や3JC’C’結合の大きさに依存する。そのため、どの程度の混合時間に設定すればよいかは、試料や磁場強度によって異なる。筆者が500MHzNMRで実験をおこなう際は、混合期間を300~500ms程度に設定し、2D版スペクトル((HN)CO(CO)(N)H、(H)N(COCO)(N)H)を測定して対角ピーク、交差ピークが十分な信号強度で得られているかを確認する。高磁場では交差ピークの信号強度が小さくて検出が困難な場合があるので、その際は混合期間を短縮して、再度2Dスペクトルを確認する。
一部のアミノ酸配列領域で横緩和時間が短い等の理由により13C’混合による磁化移動効率が悪く交差ピークが検出されない場合は、13C’混合期間を短縮したスペクトルを測定するのもよいが、次に述べるようにHN(CA)COスペクトルやHN(CA)NNHスペクトルからも残基間の相関を得ることができる。
(HN)CO(CO)NHや(H)N(COCO)NHスペクトルで交差ピークが検出できない
局所的にポリペプチド鎖がヘリックスを形成すると、(HN)CO(CO)NHスペクトルや(H)N(COCO)NHスペクトルにおいて交差ピークが検出されない場合がある。その理由として、(I)ヘリックスを形成することにより回転拡散運動が遅くなり、横緩和速度が上昇する、(II)カルボニル炭素間の3JC’C’結合はポリペプチド鎖の二面角(φ)に依存し、ヘリックスでは3JC’C’値が小さい、ことが挙げられる。このような場合には、3D HN(CA)COやHN(CA)NNHスペクトルを得ることによって13C’および15NH化学シフトの残基間の相関を得ることができる。これらの3Dスペクトルにおいても、15N核の化学シフト展開をsemi-constant timeにすることで、高分解能のスペクトルを得ることができる。
アスパラギン側鎖との相関
(H)N(COCO)NHスペクトルで、どの主鎖15NH核の化学シフト値とも一致しない信号強度の小さな交差ピークが112ppm付近にあらわれることがある。このピークは、13C’混合期間中の側鎖カルボニル炭素から主鎖への磁化移動により得られるピークである。ただし、これらのピークは側鎖アミド基の2つの水素のうち一方が重水素に置換されている場合(15NHD)にのみ検出される。すなわち、信号強度は小さく、磁場のロックのために添加する重水の濃度に依存する。また、このときの15N核の化学シフト値は重水素置換により同位体シフトしているので、1H-15NHSQCスペクトル上の側鎖信号と照らし合わせるときは注意が必要である。
試料の温度上昇
(HN)CO(CO)NHや(H)N(COCO)NH実験では、長期間の混合パルス系列やデカップリングパルスにより、測定試料の温度が上昇する場合がある。熱電対でモニターしている温度は試料内の温度と一致していないことがあるので注意が必要である。筆者が500msの混合時間での実験において、スキャン毎の水とDSSの1H化学シフト差から試料の温度上昇を見積もったところ、ダミースキャンの間に熱電対の示す温度が目標温度で一定になっていても、試料内の温度は0.5℃高いことがわかった。試料温度を厳密に調整する必要がある場合には、あらかじめ測定による温度上昇を調べ、測定時の目標温度を温度上昇する分だけ下げておく等の工夫が必要である。
蛋白質の重水素化
変性蛋白質では13C’や15NHの横緩和時間が長いため、HNCO、(HN)CO(CO)NH、(H)N(COCO)NH実験において、間接観測軸の化学シフト展開時間を長くすることにより、高分解能のスペクトルを得ることができる。その一方、直接観測する1HNの線幅は1HNと1Hαの3J結合のため、13C’や15Nの線幅と比べて広い。この問題に対して、α水素を重水素化した蛋白質を調製し、1H信号観測時間を長くすることで1HNの線幅をシャープにすることができる。このとき、15Nデカップリングによる温度上昇に注意する必要があるが、変性蛋白質では15NH核の化学シフト分散が小さいため、比較的弱いパワーでも効率よくデカップリングできる。別の方法として、信号取得中のバンド選択的同種核(BASH)デカップリング(6)によって分解能を向上することも可能であると思われる。
非線形サンプリング
高分解能3D NMRスペクトル測定では、測定に長時間を要する。試料の経時変化が問題となる場合は、非線形サンプリングをおこなうことで測定時間を短縮できる。
文献
- Wright, P.E. and Dyson, H.J., J. Mol. Biol., 293, 321-31 (1999)
- Grzesiek, S. and Bax, A., J. Biomol. NMR, 3, 185-204 (1993)
- Logan, T.M., et al., J. Biomol. NMR, 3, 225-231 (1993)
- Grzesiek, S. and Bax, A., J. Biomol. NMR, 9, 207-11 (1997)
- Yoshimura, Y., Kulminskaya, N.V., Mulder, F.M.M., J. Biomol. NMR, 61, 109-21 (2015)
- Ying, J., Roche, J., Bax, A., J. Magn. Reson., 241, 97-102 (2014)
謝辞
ここで紹介したプロトコールは、Frans A. A. Mulder准教授(オーフス大学)の指導の下で確立した方法である。本プロトコールの作成にあたり、安定同位体標識蛋白質(ユビキチン、α-シヌクレイン、FapC)を、それぞれ、Tania A. Nielsen様、Camilla B. Andersen様、Brian S. Vad博士(いずれもオーフス大学)から提供していただいた。また、EMBO Long-Term Fellowship(ALTF 687-2013)の留学支援に感謝申し上げる。
