

概要
NMR を用いたタンパク質の構造化学研究では、溶液中での立体構造決定のみならず、その動的な挙動や他のリガンド等との相互作用を原子レベルで解析することができる。一般的なタンパク質の NMR 解析では異種核多次元測定によってシグナルの縮重を解消し、かつその検出感度を向上させるために 13C および15N 同位体標識をする。この標識には、コストや技術的な側面から大腸菌組換え発現を利用する方法がおもに用いられている。よって、複雑なジスルフィド結合を形成したり糖鎖修飾をうけたりする真核生物由来のタンパク質、あるいは膜タンパク質などには適用が困難な場合がある。また解析対象が高分子量の場合、シグナルの縮重および検出感度の低下が顕著となる。そこで近年、NMR で高感度に検出が可能なメチル基などを部位特異的に安定同位体標識する手法が広まっている(1, 2)。
本稿では、部位特異的な安定同位体標識法の中でも、化学修飾によってアミノ酸側鎖に NMR 検出可能な構造プローブとしての安定同位体化合物を付加する方法について概説する。具体的には、タンパク質中のリジンおよびグルタミン側鎖への標識法とその NMR 測定に関するプロトコールを記載した。化学修飾による安定同位体標識法の利点は、タンパク質の発現系や由来を問わず標識できることや、高価な安定同位体試薬を多く用いる必要がないためコスト面ですぐれていることなどである。ただしなんらかの標識物が付加するため、標識後のタンパク質構造・物性の変化および調製したサンプルの均質度には注意をはらう必要がある。もしそれらが大きな問題とならない場合は、高分子量タンパク質(複合体)や膜タンパク質といった対象の NMR を用いた構造化学研究における有力な手法となる(3,4)。
装置・器具・試薬
1)
- 精製タンパク質(バッファーとして一級アミンである Tris は用いない)
- Dimethylamine borane: Wako #026-08402; Sigma-Aldrich #180238
- Formaldehyde (13C, 99%): Cambridge Isotope Laboratories #CLM-806-PK
- Dithiothreitol (DTT, optional)
- 脱塩カラム(PD-10: GE Healthcare #17085101; Bio-Gel P4: Bio-Rad #1504124)
- 1H-13C 検出が可能な NMR 装置
2)
- 精製タンパク質(バッファーとして一級アミンである Tris は用いない)
- Microbial transglutaminase: supplied by Ajinomoto
- 2,2,2-Trifluoroethylamine hydrochloride: TCI #T1170
- Ammonium chloride-15N: Shoko Scientific #N15-0034
- DTT
- 液体クロマトグラフィー装置(AKTAprime: GE Healthcare など)
- ゲルろ過カラム(Superdex 75 pg: GE Healthcare など)
- 1H-19F 検出が可能な NMR 装置(Bruker BBFO, BBO H&F または QCI-F プローブ)
- 1H-15N 検出が可能な NMR 装置
実験手順
- 1)リジンの 13C 標識:還元的メチル化による 13CH3 修飾
- 1-1)標識反応
- 1-2)精製
- 1-3)NMR 測定
- 2)グルタミンの 19F/15N 標識:トランスグルタミナーゼを用いた CF3 修飾 / CO15NH2 標識
- 2-1)標識反応
- 2-2)精製
- 2-3)NMR 測定
実験の詳細
1)リジンの 13C 標識:還元的メチル化による 13CH3 修飾
リジン側鎖アミノ基は中性 pH でほぼプロトン化しているものの、多くのリジン側鎖は溶媒に露出しており、わずかな非プロトン化状態が反応基となる。安定同位体標識という観点で有用な反応は、還元的メチル化である。この反応では、アミノ基とホルムアルデヒドがシッフ塩基(-N=CH2)を形成し、さらに還元されることで不可逆的にメチル(-N-CH3)化される。さらにこの二級アミンも同様にシッフ塩基を形成するため、ホルムアルデヒド過剰の条件ではジメチル(-N-(CH3)2)化される(図1A)。メチル修飾されたリジン側鎖アミノ基の pKa は通常のリジン(pKa: およそ10.5)とくらべて0.5程度しか変化しない(5)。すなわち、中性 pH 付近ではメチル化リジンも通常のリジンと同様プロトン化状態にあり、静電的性質が保持される。
中性 pH 付近で作用するシッフ塩基の還元剤として Sodium cyanoborohydride および Dimethylamine borane(Borane dimethylamine complex)が知られている(5,6)。前者は還元力が強い反面システインなどへの副反応を起こす可能性があり、後者のほうがより穏やかな還元剤として用いられる。システインへの副反応については、反応後に DTT を添加することによって付加物を除去できる場合がある(7)。
1-1)標識反応
A. 試薬溶液を調製する。
1 M Dimethylamine borane(反応前に用時調製する)
2 M Formaldehyde (13C, 99%)(CIL の試薬は約20(w/w)%水溶液であり、およそ 6.7 M となる。希釈せずにそのまま用いてもよい)
1 M DTT (optional)
pH 7-8付近のバッファー(リン酸-Na, Hepes-Na など。一級アミンである Tris は用いない)
B. 調製した試薬溶液をタンパク質溶液に加える。
ホルムアルデヒドをタンパク質中のアミノ基の50倍以上を目安に加えると、ほぼすべてのアミノ基がジメチル化される傾向にある。
C. 4℃でひと晩静置する。室温でもよい。多くの場合反応中に気泡が生じるが、問題はない。
D. 反応後に 1 M DTT を 5 μl 加える(optional)。
1-2)精製
脱塩カラムを用いて反応試薬を除く。透析や限外ろ過でもよいが、試薬が残留すると NMR 検出されるので念入りに行う。反応試薬の除去と同時に、NMR 測定用バッファーに交換する。1H-13C 相関 NMR 測定を行うためリン酸-Na バッファーが望ましい。もしくは重水素化バッファーを用いる。おなじサンプルで 1H-15N 相関 NMR 測定を行わない場合は、重水で調製したリン酸-Na バッファーまたは重水素化バッファーに交換することが望ましい。
サンプル濃度はクライオプローブでなくとも10から 25 μM 程度あれば十分である。筆者らはクライオプローブを装着した高磁場 NMR を用いて 0.2 μM(分子量: およそ35,000)でのシグナル検出に成功している(8)。また標識物の MS 測定を行っておくと、残基数に対する修飾度がおおむね同定できる。13CH3 基の付加によって分子量は15増えるため、N 末端を含むすべてのアミノ基がジメチル化された場合の分子量変化は (タンパク質中のリジンの個数 + 1 ) × (15 × 2) となる。
1-3)NMR 測定
1H-13C 検出が可能な NMR 装置であればよい。以下に軽水サンプルで N-ジメチルと N-メチルの帯域を観測する場合の、Bruker 500 MHz NMR 装置におけるセットアップ例を示す。
コマンド “rpar HSQCETGP” でパラメーターを読み込んでから “getprosol” をしてから、1H 90°パルス値(P1)の校正値をもとめたのち、
AcquPars(eda)
AcquPars(ased)
を設定する。測定時間はおよそ 25 min となる。太字以外の部分は変更しないでよい。得られるスペクトルの一例が図1Bである。以下に詳細を示す。
パルスプログラム:試料溶液を重水で調製してあり(強い)水消しが必要ない場合には、hsqcgpph または hsqcphpr を用いて States-TPPI 法で測定すると検出感度が高い。軽水溶媒の場合には hsqcetgp を用いて Echo-Antiecho 法で測定し、軽水由来のシグナルを抑制する。メチル基(I3S スピン系)の場合、sensitivity improvement(si)スキームによって期待できる感度向上はわずかであるため、si の含まれていないパルスプログラムを推奨する。
パラメーター:DSS(0 ppm)を基準として観測される化学シフトは N-ジメチル(-N-(CH3)2)が 1H: 2.8-2.9 ppm; 13C: 45-46 ppm、N-メチル(-N-CH3)が 1H: 2.7-2.8 ppm; 13C: 35-36 ppm 付近である。しかし Bruker NMR(Topspin)では 13C の基準周波数が、DSS の 1H 基準周波数に磁気回転比の比率をかけた値よりも 2.66 ppm だけ高磁場側にずれている。このため、設定値としての 13C 観測中心は N-ジメチルが 43 ppm、N-メチルが 33 ppm 付近となる。サンプルの修飾度が未知の場合は、まず 13C の観測中心 38 ppm、観測幅 20 ppm 程度で測定する。このとき N-ジメチルのシグナルのみが観測され、N-メチルのシグナルが観測されない場合は、すべてのアミノ基がジメチル化されていることを示す。N-ジメチルのシグナルのみを観測するときは 13C の観測中心 43 ppm、観測幅 10 ppm 以下に設定してもよい。
直接観測軸である 1H の設定について、通常の HSQC では 1H FID の取り込み時間(AQ)が 80-100 ms 程度になっている。リジン末端に修飾したメチル基のシグナルは比較的混み合っているが横緩和時間は長いため、取り込み時間を 200-250 ms 程度まで伸ばすことによって分解能の向上が期待できる。しかし 13C デカップルパルスは、90°パルス長換算で 60 μs(GARP-4 [figure of merit:\(\Xi = 4.8\)] での有効帯域 \({\Xi}{\gamma}B_{1}/2{\pi} = 20 \textrm{kHz}\) であり、500 MHz NMR において 13C: 160 ppm)程度に設定されている。クライオプローブでは 13C デカップル時における 1H FID 取り込み時間の推奨上限が 140 ms 程度であり、そのままの設定ではプローブへの負荷が懸念される。そこで、13C デカップルのパルス長を長くしてパルス出力を小さくする。前述のとおり観測される帯域は限られているため、デカップルの有効帯域が狭くなっても問題はない。GARP-4 ではパルス長を 240 μs まで長くしても有効帯域は 5 kHz となり、500 MHz で 40 ppm、800 MHz で 25 ppm までカバーできる。変更したパルス長でのパルス出力値(W または dB)は、コマンド “edprosol” から Square Pulses のタブを開き、13C に関するいずれかの項目に変更したいパルス長を入力すれば自動的に計算されるので、その値を 13C デカップルのパルス出力 PLW12 に入力する。もしくは、\(x_{1}\) [μsec] でのパルス出力が \(X_{1}\) [W](\(A_{1}\) [dB])のとき、\(x_{2}\) [μsec] でのパルス出力 \(X_{2}\) [W](\(A_{2}\) [dB])は、以下の関係式から求められる。
\[
\begin{align}
X_{2} [\textrm{W}] &= X_{1}\{1/(x_{2}/x_{1})\}^{2} = X_{1}(x_{1}/x_{2})^{2}\
A_{2} [\textrm{dB}] &= X_{1} + 10\log\{1/(X_{2}/X_{1})\} = A_{1} - 20\log(x_{1}/x_{2})
\end{align}
\]
以上より簡単には、パルス長が2倍になればパルス出力は\(1/4\) [W](\(+6\) [dB])になる。
2)グルタミンの 19F/15N 標識:トランスグルタミナーゼを用いた CF3 修飾 / CO15NH2 標識
本来、グルタミン側鎖カルバミドは化学的に安定であり、化学修飾反応の標的とはならない。ところがトランスグルタミナーゼとよばれる酵素を用いた触媒反応を利用することにより、修飾反応が可能となる。トランスグルタミナーゼはグルタミン側鎖とリジン側鎖間のイソペプチド結合による架橋反応を触媒する。この活性を利用すれば、グルタミン側鎖にリジン様の一級アミン化合物を付加することができる。
筆者らはその基質として 2,2,2-Trifluoroethylamine hydrochloride を用いることで、グルタミン側鎖の CF3 修飾に成功した(図2A)(9)。また以前には、基質として Ammonium Chloride-15N を用いた側鎖カルバミド(-CONH2)の 15N 標識法が報告されている(図3A)(10)。15N 標識法ではグルタミン側鎖の化学構造を変化させないため、本来のタンパク質構造・物性には影響を与えない。しかし、側鎖カルバミドの2つの 1H シグナルは別々のピークとして観測される一方、CF3 では3原子分の 19F シグナルが1つのピークとして縮重するため、検出感度において優位性がある。
トランスグルタミナーゼを用いた方法は酵素触媒反応であるため、タンパク質中のどのグルタミン残基にどの程度の効率で標識されるかは、標的とするタンパク質の性状や標識部位近傍の立体構造に依存する。タンパク質に内在するグルタミンが基質となるには、まず酵素の基質ポケットに認識されやすい表面残基であることが必要条件となる。つぎに、標識部位近傍の構造がフレキシブルである(disorder している)と基質になりやすい(9,11)。また、結晶構造の B ファクターやプロテアーゼによる切断部位との関連も報告されている(12)。ところが最近になって報告された Protein G B1 ドメインを用いた研究では、ヘリックスやストランド上に変異導入したグルタミンの一部が反応性の高い基質となっており(13)、その基質認識様式には未解明の点がある。
2-1)標識反応
A.反応用試薬を準備する。
2 mg/ml Microbial transglutaminase
0.5 M 2,2,2-Trifluoroethylamine hydrochloride(19F 標識用)
2 M Ammonium Chloride-15N(15N 標識用)
1 M DTT
pH 7-8付近のバッファー(リン酸-Na, Hepes-Na など。一級アミンである Tris は用いない)
B. 以下の濃度になるよう、タンパク質に試薬を加える。
目的とする安定同位体標識(19F 標識または15N 標識)に応じていずれかの基質化合物を加える。19F 標識の場合、基質となるタンパク質によっては修飾物が変性または沈殿することがある。また、2,2,2-Trifluoroethylamine hydrochloride の添加によって pH が下がる場合があり、問題となるようなら試薬濃度やバッファー濃度を検討する。もしくはあらかじめストック溶液の pH を調整しておく。DTT はトランスグルタミナーゼの活性中心にあるシステインを還元状態に保つ目的で加える。標的タンパク質がジスルフィド結合によって構造形成している場合は加えない。
トランスグルタミナーゼは味の素㈱で調製された試薬を用いている。くわしい入手方法については問い合わせされたい。同等の試薬としては、Zedira という海外メーカーから市販されている。Sigma-Aldrich から市販のトランスグルタミナーゼは guinea pig liver(モルモット肝臓)由来であり、Microbial(Streptomyces mobaraensis)由来のトランスグルタミナーゼよりも基質特異性が高い(基質となるグルタミンが限定される)。
C. 室温(20-25℃)でひと晩静置する。なおトランスグルタミナーゼは37℃付近でもっとも活性が高いため、熱安定性の高いタンパク質であれば、37℃, 3 h といった条件で反応させてもよい。
2-2)精製
ゲルろ過カラムでトランスグルタミナーゼと反応試薬を除く。トランスグルタミナーゼ(分子量: およそ38,000)と目的タンパク質の分離が難しい場合は、イオン交換カラムなどで精製するか、標的タンパク質をアフィニティータグとの融合体のまま反応させ、アフィニティーカラムで標的タンパク質とトランスグルタミナーゼを分離する。
サンプル濃度は、クライオプローブでなくとも10から 50 μM 程度あれば十分である。NMR 測定用バッファーは、19F 標識サンプルの場合とくに制約はない。15N 標識サンプルでは通常の 1H-15N HSQC 用サンプルの場合と同様に、軽水溶媒で調製する。
2-3)NMR 測定
19F 標識サンプルの場合、1H-19F 相関 NMR シグナルの検出が可能な NMR 装置であればよい。CH2 との J カップリングがあるため、19F FID 取り込み時に 1H デカップルをしないとピーク分裂する。BBFO または BBO H&F(1H/19F,X)もしくはQCI-F(1H/13C/15N/19F)プローブでは1H デカップルが可能だが、BBO(1H,19F/X)プローブでは 1H と19F が共通チャンネルのため 1H デカップルできない。以下に Bruker 400 MHz NMR 装置におけるセットアップ例を示す。
コマンド “rpar F19CPD” でパラメーターを読み込んだのち、
AcquPars(eda)
AcquPars(ased)
を設定する。測定時間はおよそ 25 min となる。太字以外の部分は変更しないでよい。得られるスペクトルの一例が図2Bである。以下に詳細を示す。
パルスプログラム:1H デカップルのために zgfhigqn.2 を用いる。zgflqn(パラメーター: F19)には 1H デカップルが含まれていないため注意する。
パラメーター:グルタミン側鎖に修飾された (-CONH)CH2CF3 の 19F シグナルは -72 ppm 付近に検出される。90°パルス設定はコマンド “getprosol” で読み込んだデフォルト値を用いてもよいが、1H 核の場合と同様にバッファーの塩濃度によっては90°パルス長が長くなる。よって、デフォルト値から 0.5 μsec 程度の間隔で値を大きくしていき、目的とするシグナル強度が最大となる値に設定する。もしくは TFA を internal または external リファレンスとして測定し、パルス長校正および化学シフト校正(-75.39 ppm)に利用する。
19F FID の取り込み時間はデフォルトで 500-600 ms 程度であるが、筆者が調製したサンプルでは 250-300 ms 程度で感度と分解能のバランスがいいように思われた。また標識部位の運動性や化学交換に依存するが、ピークの線幅が広がりやすい(>4 Hz)傾向にあるため、exponential 型のウィンドウ関数(EM)の Line broadening(LB)をそれにあわせて 4 Hz 程度に設定している。ただし、19F シグナルはとくに高磁場でブロードニングしやすい傾向があるため、実際の線幅やピーク数、検出感度に応じて変更する。
15N 標識サンプルの場合、1H-15N 相関 NMR 検出が可能な NMR 装置であればよい。測定は一般的な 1H-15N 相関を検出する WATERGATE-HSQC のパルスプログラムを用いる。得られるスペクトルの一例が図3Bである。ピーク数が少なく 15N 軸方向への分離を必要としない場合は、15N-edited 1H 1D 測定でもよい。1D 測定にするには AcquPars にある F1(15N) の TD を1にするか、↓1,2,..のアイコン(Change acquisition dimension of current data set)をクリックして 1D に変更する。
工夫とコツ
帰属
化学修飾による安定同位体標識サンプルのシグナル帰属は、遺伝子変異によるアミノ酸置換によって行う。リジンの場合はアルギニンに、グルタミンの場合はアスパラギンに置換する。溶媒露出度が高い残基であれば、変異した残基に由来するピークのみが消失し、その他のピークはほとんど変化しない。一方で溶媒露出度が低い、あるいは他の残基との相互作用がある残基では、ピーク消失とあわせて他のピークに変化がみられる場合がある。しかし立体構造が既知であるならば、そのような変異導入によるスペクトル変化の関係性は、帰属を裏づける情報となる。
トランスグルタミナーゼを用いた標識反応では、変異導入によるわずかな構造変化によって修飾反応への特異性に影響を与え、想定外のスペクトル変化がまれに起こる。また表面露出度が極めて高い場合、アスパラギンであっても基質となる場合がある(図3B)(9)。
測定温度
リジンの 13CH3 修飾およびグルタミンの CF3 修飾においては、アミノ酸側鎖の運動性がピーク線形に大きく影響するため、測定温度は重要である。リジンの 13CH3 修飾ではおもにジメチル(-N-(CH3)2)化されるため、その回転が遅くなると2つのメチル基の化学交換速度が低下し、ピークが分裂あるいはブロードニングする(図1B)。逆に回転が十分に速い場合は平均化され、1つのピークとして観測される。よってスペクトルを簡略化するには、回転運動を速めるために高温(>40℃)で測定するほうがよい(14)。室温付近(20-30℃)ではリジン側鎖の相互作用・運動性に応じて、1つのシャープまたはブロードなピーク、および分裂したピークとして観測される(8,14)。そのようなピーク分裂あるいはブロードニングは、酸性アミノ酸残基との塩橋形成によってジメチルの回転運動がおさえられることに起因する場合が多い(4,8,15)。
トランスグルタミナーゼによるグルタミンの CF3 修飾では、カルバミドの平面性に起因するシストランスの化学交換が存在する。CF3 はカルバミドの N 原子から2結合分(-CONHCH2CF3)へだてているためその影響は顕著ではないが、低い温度ではピーク分裂やブロードニングが懸念されるため、推奨しない。筆者らは30℃の測定にて1本のピークとして観測されている(図2B)。
測定磁場
化学交換の速度が同じであっても化学交換している共鳴周波数(Hz)差が変化するため、測定磁場も重要である。低磁場では共鳴周波数差が小さくなり、速い交換状態(fast exchange)にあるスペクトルが得られやすい。高磁場では逆に共鳴周波数差が大きくなり、遅い交換状態(slow exchange)となりやすい。すなわち高温・低磁場では fast exchange よりに、低温・高磁場では slow exchange よりになる。
またグルタミンの CF3修飾における 19F-NMR 測定では、多核 NMR に特有の大きな化学シフト異方性による緩和の影響が顕著であり、高磁場化による分解能・感度向上は 1H-NMR ほど期待できない。
測定 pH
リジンの 13CH3 修飾においては、アミノ基のプロトン化状態およびそのプロトン交換速度がメチル基の化学シフトおよびピーク線形に影響をあたえる。pH を塩基性(pH >10)にするとアミノ基のプロトンが解離し、1H 化学シフトは 0.7 ppm ほど高磁場側に、13C 化学シフトは 1 ppm ほど低磁場側にシフトする(16)。ジメチルアミノ基の pKa はおよそ10であるため、もし pH 10 付近での測定が可能であるならば、残基ごとの pKa の違いにしたがって混み合ったピークを分散させることができる。また、pH をやや酸性より(pH <6)にするとアミノ基のプロトン交換速度が低下することに由来して、ピークが分裂またはブロードニングする場合がある(15–17)。
スペクトル変化の解析
分子間相互作用等にともなう構造変化をスペクトル変化から解析する場合、化学修飾によって導入した安定同位体標識部位はアミノ酸側鎖の末端であるため、化学シフト変化量自体は小さいことが多い。しかしながら、スペクトル全体からみた相対的な変化量は微細な化学環境の変化を鋭敏に反映する。リジンの 13CH3 修飾の場合では、1H 化学シフト変化量にして 0.01 ppm が、ひとつのしきい値となる(8)。13C 化学シフト変化も指標となりうるが相対的な変化量は 1H とくらべて小さく、また上記で述べた化学交換の影響をうけやすいため、1H 変化量のみを解析するほうが簡便である。グルタミンの CF3 修飾の場合、0.1 ppm 程度が有意な 19F 化学シフト変化量として期待される(9)。さらに、19F 核を含む多核に特有の広い化学シフト範囲および大きな化学シフト異方性によって、ピークブロードニングが引き起こされやすい傾向にある。
修飾反応の影響
実験の詳細1)で示したように、リジンの 13CH3 修飾では側鎖アミノ基の pKa が大きく変わらないため、中性 pH 付近ではプロトン化して正に帯電する。よって、酸性アミノ酸残基との塩橋といった静電的相互作用も保持されると考えられる。筆者らの研究では、メチル修飾した3種のタンパク質についての系統的な解析から、遅い化学交換によるピーク分裂と塩橋形成が強い相関を示し、逆に速い化学交換によって1本のピークとして観測される場合には露出した表面残基であることが示された(18)。またメチル修飾による立体構造変化は主鎖原子の RMSD にして 1 Å 以内であり、全体としてのフォールディングは保持される(8)。筆者らが NMR によって構造決定した FKBP12 のリジンメチル修飾体(PDB: 2ND5)では、非修飾体との主鎖 RMSD が 0.8 Å であった(18)。ただし、タンパク質の結晶化においてはリジンメチル修飾による格子間のパッキング効果によって結晶化能や分解能の向上がみられるように(19)、タンパク質の物性になにかしらの影響が起きることは考えうる。また、立体構造への影響という観点では表面残基よりも内部残基への修飾のほうが顕著である。そこで添加するホルムアルデヒドを10から 20 mM 程度に減らす、あるいは2, 3回に分けて試薬を添加するなど、修飾反応条件を最適化することによって内部残基への修飾をおさえることができる(8)。
トランスグルタミナーゼを用いたグルタミンの標識では、15N 標識の場合には化学構造が保持される。一方の 19F 標識における CF3 修飾では表面残基の疎水性が上がり、溶解度といった性状に影響を与え、場合によっては反応中に沈殿物が生じることがある。そのような場合は、2,2,2-Trifluoroethylamine hydrochloride の濃度や反応温度を下げるなどして、条件検討する。ただし沈殿物が生じた場合であっても、ある特定の残基が修飾された場合にのみ沈殿することがあるため、反応液の上清から他の残基がCF3修飾されたサンプルを得られることがある。
システインへの化学修飾
本稿ではリジンとグルタミンへの化学修飾について概説したが、部位選択的標識という観点ではシステインへの化学修飾がもっとも有力な手段となる。とりわけ CF3 修飾による 19F 標識は GPCR への応用が報告されるなど(20, 21)、化学修飾による安定同位体標識法において重要な位置をしめてきている。また安定同位体標識のほかにも常磁性タグ(スピンラベル)標識によって、NMR では常磁性緩和促進(PRE)や擬コンタクトシフト(PCS)といった常磁性効果による解析が、さらには電子スピン共鳴(EPR)への応用が可能となる。
実験の安全
リジンの還元的メチル化反応において Sodium cyanoborohydride を還元剤として用いる場合は、毒物及び劇物取締法に係るシアン化物であるため、手袋等で防護の上で取扱い、廃棄にあたっては法令にしたがう。
文献
- Wiesner, S. & Sprangers, R., Curr. Opin. Struct. Biol., 35, 60–67 (2015)
- Zhang, H. & van Ingen, H., Curr. Opin. Struct. Biol., 38, 75–82 (2016)
- Religa, T. L. et al., J. Am. Chem. Soc., 133, 9063–9068 (2011)
- Bokoch, M. P. et al., Nature, 463, 108–112 (2010)
- Jentoft, J. E. et al., J. Biol. Chem., 254, 4366–4370 (1979)
- Cabacungan, J. C. et al., Anal. Biochem., 124, 272–278 (1982)
- Rayment, I., Methods Enzymol., 276, 171–179 (1997)
- Hattori, Y. et al., J. Biomol. NMR, 55, 19–31 (2013)
- Hattori, Y. et al., J. Biomol. NMR, 68, 271–279 (2017)
- Shimba, N. et al., Anal. Biochem., 301, 123–127 (2002)
- Spolaore, B. et al., Biochemistry, 51, 8679–8689 (2012)
- Fontana, A. et al., Adv. Drug Deliver. Rev., 60, 130–28 (2008)
- Rachel, N. M. et al., Protein Sci., 26, 2268–2279 (2017)
- 服部 良一,児嶋 長次郎,生物物理,56,288–289 (2016)
- Gerken, T. A. et al., J. Biol. Chem., 257, 2894–2900 (1982)
- Zhang, M. & Vogel, H. J., J. Biol. Chem., 268, 22420–22428 (1993)
- Larda, S. T., et al., J. Biomol. NMR, 54, 199–209 (2012)
- Hattori, Y. et al., XXVIIth ICMRBS, Kyoto, Poster046-Mon (2016)
- Walter, T. S. et al., Structure, 14, 1617–1622 (2006)
- Liu, A. et al., Science, 335, 1106–1110 (2012)
- Manglik, A. et al., Cell, 161, 1101–1111 (2015)
-

図1:(A)リジン側鎖の還元的メチル化による 13CH3 修飾反応(DMAB: Dimethylamine borane)。(B)リジン側鎖および N 末端が 13C-ジメチル化されたタンパク質 FKBP12(分子量: およそ12,000; リジン: 8残基および N 末端)の2D 1H-13C HSQC NMR スペクトル(400 MHz, 303 K)。 -
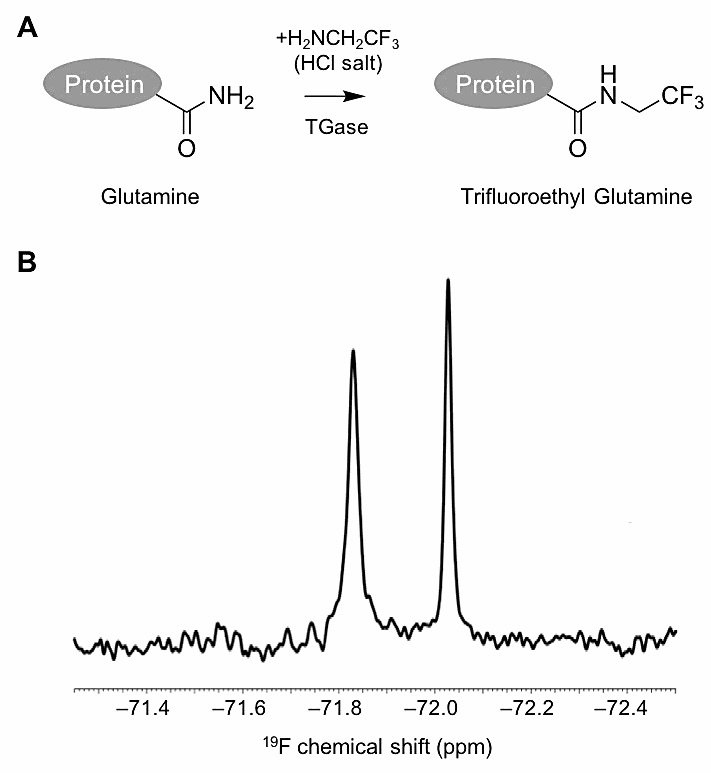
図2:(A)グルタミン側鎖のトランスグルタミナーゼ(TGase)を用いた CF3 修飾反応。(B)一部のグルタミン側鎖が CF3 修飾されたタンパク質 FKBP12(分子量: およそ12,000; グルタミン: 5残基)の1D 19F NMR スペクトル(400 MHz, 303 K)。 -

図3:(A)グルタミン側鎖のトランスグルタミナーゼ(TGase)を用いた 15N 標識反応。(B)グルタミン(およびアスパラギン)側鎖が 15N 標識されたタンパク質 FKBP12(分子量: 12,000; グルタミン: 5残基; アスパラギン: 1残基)の 2D 1H-15N HSQC NMR スペクトル(600 MHz, 303 K)。
概要
NMR を用いたタンパク質の構造化学研究では、溶液中での立体構造決定のみならず、その動的な挙動や他のリガンド等との相互作用を原子レベルで解析することができる。一般的なタンパク質の NMR 解析では異種核多次元測定によってシグナルの縮重を解消し、かつその検出感度を向上させるために 13C および15N 同位体標識をする。この標識には、コストや技術的な側面から大腸菌組換え発現を利用する方法がおもに用いられている。よって、複雑なジスルフィド結合を形成したり糖鎖修飾をうけたりする真核生物由来のタンパク質、あるいは膜タンパク質などには適用が困難な場合がある。また解析対象が高分子量の場合、シグナルの縮重および検出感度の低下が顕著となる。そこで近年、NMR で高感度に検出が可能なメチル基などを部位特異的に安定同位体標識する手法が広まっている(1, 2)。
本稿では、部位特異的な安定同位体標識法の中でも、化学修飾によってアミノ酸側鎖に NMR 検出可能な構造プローブとしての安定同位体化合物を付加する方法について概説する。具体的には、タンパク質中のリジンおよびグルタミン側鎖への標識法とその NMR 測定に関するプロトコールを記載した。化学修飾による安定同位体標識法の利点は、タンパク質の発現系や由来を問わず標識できることや、高価な安定同位体試薬を多く用いる必要がないためコスト面ですぐれていることなどである。ただしなんらかの標識物が付加するため、標識後のタンパク質構造・物性の変化および調製したサンプルの均質度には注意をはらう必要がある。もしそれらが大きな問題とならない場合は、高分子量タンパク質(複合体)や膜タンパク質といった対象の NMR を用いた構造化学研究における有力な手法となる(3,4)。
装置・器具・試薬
1)
- 精製タンパク質(バッファーとして一級アミンである Tris は用いない)
- Dimethylamine borane: Wako #026-08402; Sigma-Aldrich #180238
- Formaldehyde (13C, 99%): Cambridge Isotope Laboratories #CLM-806-PK
- Dithiothreitol (DTT, optional)
- 脱塩カラム(PD-10: GE Healthcare #17085101; Bio-Gel P4: Bio-Rad #1504124)
- 1H-13C 検出が可能な NMR 装置
2)
- 精製タンパク質(バッファーとして一級アミンである Tris は用いない)
- Microbial transglutaminase: supplied by Ajinomoto
- 2,2,2-Trifluoroethylamine hydrochloride: TCI #T1170
- Ammonium chloride-15N: Shoko Scientific #N15-0034
- DTT
- 液体クロマトグラフィー装置(AKTAprime: GE Healthcare など)
- ゲルろ過カラム(Superdex 75 pg: GE Healthcare など)
- 1H-19F 検出が可能な NMR 装置(Bruker BBFO, BBO H&F または QCI-F プローブ)
- 1H-15N 検出が可能な NMR 装置
実験手順
- 1)リジンの 13C 標識:還元的メチル化による 13CH3 修飾
- 1-1)標識反応
- 1-2)精製
- 1-3)NMR 測定
- 2)グルタミンの 19F/15N 標識:トランスグルタミナーゼを用いた CF3 修飾 / CO15NH2 標識
- 2-1)標識反応
- 2-2)精製
- 2-3)NMR 測定
実験の詳細
1)リジンの 13C 標識:還元的メチル化による 13CH3 修飾
リジン側鎖アミノ基は中性 pH でほぼプロトン化しているものの、多くのリジン側鎖は溶媒に露出しており、わずかな非プロトン化状態が反応基となる。安定同位体標識という観点で有用な反応は、還元的メチル化である。この反応では、アミノ基とホルムアルデヒドがシッフ塩基(-N=CH2)を形成し、さらに還元されることで不可逆的にメチル(-N-CH3)化される。さらにこの二級アミンも同様にシッフ塩基を形成するため、ホルムアルデヒド過剰の条件ではジメチル(-N-(CH3)2)化される(図1A)。メチル修飾されたリジン側鎖アミノ基の pKa は通常のリジン(pKa: およそ10.5)とくらべて0.5程度しか変化しない(5)。すなわち、中性 pH 付近ではメチル化リジンも通常のリジンと同様プロトン化状態にあり、静電的性質が保持される。
中性 pH 付近で作用するシッフ塩基の還元剤として Sodium cyanoborohydride および Dimethylamine borane(Borane dimethylamine complex)が知られている(5,6)。前者は還元力が強い反面システインなどへの副反応を起こす可能性があり、後者のほうがより穏やかな還元剤として用いられる。システインへの副反応については、反応後に DTT を添加することによって付加物を除去できる場合がある(7)。
1-1)標識反応
A. 試薬溶液を調製する。
1 M Dimethylamine borane(反応前に用時調製する)
2 M Formaldehyde (13C, 99%)(CIL の試薬は約20(w/w)%水溶液であり、およそ 6.7 M となる。希釈せずにそのまま用いてもよい)
1 M DTT (optional)
pH 7-8付近のバッファー(リン酸-Na, Hepes-Na など。一級アミンである Tris は用いない)
B. 調製した試薬溶液をタンパク質溶液に加える。
ホルムアルデヒドをタンパク質中のアミノ基の50倍以上を目安に加えると、ほぼすべてのアミノ基がジメチル化される傾向にある。
C. 4℃でひと晩静置する。室温でもよい。多くの場合反応中に気泡が生じるが、問題はない。
D. 反応後に 1 M DTT を 5 μl 加える(optional)。
1-2)精製
脱塩カラムを用いて反応試薬を除く。透析や限外ろ過でもよいが、試薬が残留すると NMR 検出されるので念入りに行う。反応試薬の除去と同時に、NMR 測定用バッファーに交換する。1H-13C 相関 NMR 測定を行うためリン酸-Na バッファーが望ましい。もしくは重水素化バッファーを用いる。おなじサンプルで 1H-15N 相関 NMR 測定を行わない場合は、重水で調製したリン酸-Na バッファーまたは重水素化バッファーに交換することが望ましい。
サンプル濃度はクライオプローブでなくとも10から 25 μM 程度あれば十分である。筆者らはクライオプローブを装着した高磁場 NMR を用いて 0.2 μM(分子量: およそ35,000)でのシグナル検出に成功している(8)。また標識物の MS 測定を行っておくと、残基数に対する修飾度がおおむね同定できる。13CH3 基の付加によって分子量は15増えるため、N 末端を含むすべてのアミノ基がジメチル化された場合の分子量変化は (タンパク質中のリジンの個数 + 1 ) × (15 × 2) となる。
1-3)NMR 測定
1H-13C 検出が可能な NMR 装置であればよい。以下に軽水サンプルで N-ジメチルと N-メチルの帯域を観測する場合の、Bruker 500 MHz NMR 装置におけるセットアップ例を示す。
コマンド “rpar HSQCETGP” でパラメーターを読み込んでから “getprosol” をしてから、1H 90°パルス値(P1)の校正値をもとめたのち、
AcquPars(eda)
AcquPars(ased)
を設定する。測定時間はおよそ 25 min となる。太字以外の部分は変更しないでよい。得られるスペクトルの一例が図1Bである。以下に詳細を示す。
パルスプログラム:試料溶液を重水で調製してあり(強い)水消しが必要ない場合には、hsqcgpph または hsqcphpr を用いて States-TPPI 法で測定すると検出感度が高い。軽水溶媒の場合には hsqcetgp を用いて Echo-Antiecho 法で測定し、軽水由来のシグナルを抑制する。メチル基(I3S スピン系)の場合、sensitivity improvement(si)スキームによって期待できる感度向上はわずかであるため、si の含まれていないパルスプログラムを推奨する。
パラメーター:DSS(0 ppm)を基準として観測される化学シフトは N-ジメチル(-N-(CH3)2)が 1H: 2.8-2.9 ppm; 13C: 45-46 ppm、N-メチル(-N-CH3)が 1H: 2.7-2.8 ppm; 13C: 35-36 ppm 付近である。しかし Bruker NMR(Topspin)では 13C の基準周波数が、DSS の 1H 基準周波数に磁気回転比の比率をかけた値よりも 2.66 ppm だけ高磁場側にずれている。このため、設定値としての 13C 観測中心は N-ジメチルが 43 ppm、N-メチルが 33 ppm 付近となる。サンプルの修飾度が未知の場合は、まず 13C の観測中心 38 ppm、観測幅 20 ppm 程度で測定する。このとき N-ジメチルのシグナルのみが観測され、N-メチルのシグナルが観測されない場合は、すべてのアミノ基がジメチル化されていることを示す。N-ジメチルのシグナルのみを観測するときは 13C の観測中心 43 ppm、観測幅 10 ppm 以下に設定してもよい。
直接観測軸である 1H の設定について、通常の HSQC では 1H FID の取り込み時間(AQ)が 80-100 ms 程度になっている。リジン末端に修飾したメチル基のシグナルは比較的混み合っているが横緩和時間は長いため、取り込み時間を 200-250 ms 程度まで伸ばすことによって分解能の向上が期待できる。しかし 13C デカップルパルスは、90°パルス長換算で 60 μs(GARP-4 [figure of merit:\(\Xi = 4.8\)] での有効帯域 \({\Xi}{\gamma}B_{1}/2{\pi} = 20 \textrm{kHz}\) であり、500 MHz NMR において 13C: 160 ppm)程度に設定されている。クライオプローブでは 13C デカップル時における 1H FID 取り込み時間の推奨上限が 140 ms 程度であり、そのままの設定ではプローブへの負荷が懸念される。そこで、13C デカップルのパルス長を長くしてパルス出力を小さくする。前述のとおり観測される帯域は限られているため、デカップルの有効帯域が狭くなっても問題はない。GARP-4 ではパルス長を 240 μs まで長くしても有効帯域は 5 kHz となり、500 MHz で 40 ppm、800 MHz で 25 ppm までカバーできる。変更したパルス長でのパルス出力値(W または dB)は、コマンド “edprosol” から Square Pulses のタブを開き、13C に関するいずれかの項目に変更したいパルス長を入力すれば自動的に計算されるので、その値を 13C デカップルのパルス出力 PLW12 に入力する。もしくは、\(x_{1}\) [μsec] でのパルス出力が \(X_{1}\) [W](\(A_{1}\) [dB])のとき、\(x_{2}\) [μsec] でのパルス出力 \(X_{2}\) [W](\(A_{2}\) [dB])は、以下の関係式から求められる。
\[
\begin{align}
X_{2} [\textrm{W}] &= X_{1}\{1/(x_{2}/x_{1})\}^{2} = X_{1}(x_{1}/x_{2})^{2}\
A_{2} [\textrm{dB}] &= X_{1} + 10\log\{1/(X_{2}/X_{1})\} = A_{1} - 20\log(x_{1}/x_{2})
\end{align}
\]
以上より簡単には、パルス長が2倍になればパルス出力は\(1/4\) [W](\(+6\) [dB])になる。
2)グルタミンの 19F/15N 標識:トランスグルタミナーゼを用いた CF3 修飾 / CO15NH2 標識
本来、グルタミン側鎖カルバミドは化学的に安定であり、化学修飾反応の標的とはならない。ところがトランスグルタミナーゼとよばれる酵素を用いた触媒反応を利用することにより、修飾反応が可能となる。トランスグルタミナーゼはグルタミン側鎖とリジン側鎖間のイソペプチド結合による架橋反応を触媒する。この活性を利用すれば、グルタミン側鎖にリジン様の一級アミン化合物を付加することができる。
筆者らはその基質として 2,2,2-Trifluoroethylamine hydrochloride を用いることで、グルタミン側鎖の CF3 修飾に成功した(図2A)(9)。また以前には、基質として Ammonium Chloride-15N を用いた側鎖カルバミド(-CONH2)の 15N 標識法が報告されている(図3A)(10)。15N 標識法ではグルタミン側鎖の化学構造を変化させないため、本来のタンパク質構造・物性には影響を与えない。しかし、側鎖カルバミドの2つの 1H シグナルは別々のピークとして観測される一方、CF3 では3原子分の 19F シグナルが1つのピークとして縮重するため、検出感度において優位性がある。
トランスグルタミナーゼを用いた方法は酵素触媒反応であるため、タンパク質中のどのグルタミン残基にどの程度の効率で標識されるかは、標的とするタンパク質の性状や標識部位近傍の立体構造に依存する。タンパク質に内在するグルタミンが基質となるには、まず酵素の基質ポケットに認識されやすい表面残基であることが必要条件となる。つぎに、標識部位近傍の構造がフレキシブルである(disorder している)と基質になりやすい(9,11)。また、結晶構造の B ファクターやプロテアーゼによる切断部位との関連も報告されている(12)。ところが最近になって報告された Protein G B1 ドメインを用いた研究では、ヘリックスやストランド上に変異導入したグルタミンの一部が反応性の高い基質となっており(13)、その基質認識様式には未解明の点がある。
2-1)標識反応
A.反応用試薬を準備する。
2 mg/ml Microbial transglutaminase
0.5 M 2,2,2-Trifluoroethylamine hydrochloride(19F 標識用)
2 M Ammonium Chloride-15N(15N 標識用)
1 M DTT
pH 7-8付近のバッファー(リン酸-Na, Hepes-Na など。一級アミンである Tris は用いない)
B. 以下の濃度になるよう、タンパク質に試薬を加える。
目的とする安定同位体標識(19F 標識または15N 標識)に応じていずれかの基質化合物を加える。19F 標識の場合、基質となるタンパク質によっては修飾物が変性または沈殿することがある。また、2,2,2-Trifluoroethylamine hydrochloride の添加によって pH が下がる場合があり、問題となるようなら試薬濃度やバッファー濃度を検討する。もしくはあらかじめストック溶液の pH を調整しておく。DTT はトランスグルタミナーゼの活性中心にあるシステインを還元状態に保つ目的で加える。標的タンパク質がジスルフィド結合によって構造形成している場合は加えない。
トランスグルタミナーゼは味の素㈱で調製された試薬を用いている。くわしい入手方法については問い合わせされたい。同等の試薬としては、Zedira という海外メーカーから市販されている。Sigma-Aldrich から市販のトランスグルタミナーゼは guinea pig liver(モルモット肝臓)由来であり、Microbial(Streptomyces mobaraensis)由来のトランスグルタミナーゼよりも基質特異性が高い(基質となるグルタミンが限定される)。
C. 室温(20-25℃)でひと晩静置する。なおトランスグルタミナーゼは37℃付近でもっとも活性が高いため、熱安定性の高いタンパク質であれば、37℃, 3 h といった条件で反応させてもよい。
2-2)精製
ゲルろ過カラムでトランスグルタミナーゼと反応試薬を除く。トランスグルタミナーゼ(分子量: およそ38,000)と目的タンパク質の分離が難しい場合は、イオン交換カラムなどで精製するか、標的タンパク質をアフィニティータグとの融合体のまま反応させ、アフィニティーカラムで標的タンパク質とトランスグルタミナーゼを分離する。
サンプル濃度は、クライオプローブでなくとも10から 50 μM 程度あれば十分である。NMR 測定用バッファーは、19F 標識サンプルの場合とくに制約はない。15N 標識サンプルでは通常の 1H-15N HSQC 用サンプルの場合と同様に、軽水溶媒で調製する。
2-3)NMR 測定
19F 標識サンプルの場合、1H-19F 相関 NMR シグナルの検出が可能な NMR 装置であればよい。CH2 との J カップリングがあるため、19F FID 取り込み時に 1H デカップルをしないとピーク分裂する。BBFO または BBO H&F(1H/19F,X)もしくはQCI-F(1H/13C/15N/19F)プローブでは1H デカップルが可能だが、BBO(1H,19F/X)プローブでは 1H と19F が共通チャンネルのため 1H デカップルできない。以下に Bruker 400 MHz NMR 装置におけるセットアップ例を示す。
コマンド “rpar F19CPD” でパラメーターを読み込んだのち、
AcquPars(eda)
AcquPars(ased)
を設定する。測定時間はおよそ 25 min となる。太字以外の部分は変更しないでよい。得られるスペクトルの一例が図2Bである。以下に詳細を示す。
パルスプログラム:1H デカップルのために zgfhigqn.2 を用いる。zgflqn(パラメーター: F19)には 1H デカップルが含まれていないため注意する。
パラメーター:グルタミン側鎖に修飾された (-CONH)CH2CF3 の 19F シグナルは -72 ppm 付近に検出される。90°パルス設定はコマンド “getprosol” で読み込んだデフォルト値を用いてもよいが、1H 核の場合と同様にバッファーの塩濃度によっては90°パルス長が長くなる。よって、デフォルト値から 0.5 μsec 程度の間隔で値を大きくしていき、目的とするシグナル強度が最大となる値に設定する。もしくは TFA を internal または external リファレンスとして測定し、パルス長校正および化学シフト校正(-75.39 ppm)に利用する。
19F FID の取り込み時間はデフォルトで 500-600 ms 程度であるが、筆者が調製したサンプルでは 250-300 ms 程度で感度と分解能のバランスがいいように思われた。また標識部位の運動性や化学交換に依存するが、ピークの線幅が広がりやすい(>4 Hz)傾向にあるため、exponential 型のウィンドウ関数(EM)の Line broadening(LB)をそれにあわせて 4 Hz 程度に設定している。ただし、19F シグナルはとくに高磁場でブロードニングしやすい傾向があるため、実際の線幅やピーク数、検出感度に応じて変更する。
15N 標識サンプルの場合、1H-15N 相関 NMR 検出が可能な NMR 装置であればよい。測定は一般的な 1H-15N 相関を検出する WATERGATE-HSQC のパルスプログラムを用いる。得られるスペクトルの一例が図3Bである。ピーク数が少なく 15N 軸方向への分離を必要としない場合は、15N-edited 1H 1D 測定でもよい。1D 測定にするには AcquPars にある F1(15N) の TD を1にするか、↓1,2,..のアイコン(Change acquisition dimension of current data set)をクリックして 1D に変更する。
工夫とコツ
帰属
化学修飾による安定同位体標識サンプルのシグナル帰属は、遺伝子変異によるアミノ酸置換によって行う。リジンの場合はアルギニンに、グルタミンの場合はアスパラギンに置換する。溶媒露出度が高い残基であれば、変異した残基に由来するピークのみが消失し、その他のピークはほとんど変化しない。一方で溶媒露出度が低い、あるいは他の残基との相互作用がある残基では、ピーク消失とあわせて他のピークに変化がみられる場合がある。しかし立体構造が既知であるならば、そのような変異導入によるスペクトル変化の関係性は、帰属を裏づける情報となる。
トランスグルタミナーゼを用いた標識反応では、変異導入によるわずかな構造変化によって修飾反応への特異性に影響を与え、想定外のスペクトル変化がまれに起こる。また表面露出度が極めて高い場合、アスパラギンであっても基質となる場合がある(図3B)(9)。
測定温度
リジンの 13CH3 修飾およびグルタミンの CF3 修飾においては、アミノ酸側鎖の運動性がピーク線形に大きく影響するため、測定温度は重要である。リジンの 13CH3 修飾ではおもにジメチル(-N-(CH3)2)化されるため、その回転が遅くなると2つのメチル基の化学交換速度が低下し、ピークが分裂あるいはブロードニングする(図1B)。逆に回転が十分に速い場合は平均化され、1つのピークとして観測される。よってスペクトルを簡略化するには、回転運動を速めるために高温(>40℃)で測定するほうがよい(14)。室温付近(20-30℃)ではリジン側鎖の相互作用・運動性に応じて、1つのシャープまたはブロードなピーク、および分裂したピークとして観測される(8,14)。そのようなピーク分裂あるいはブロードニングは、酸性アミノ酸残基との塩橋形成によってジメチルの回転運動がおさえられることに起因する場合が多い(4,8,15)。
トランスグルタミナーゼによるグルタミンの CF3 修飾では、カルバミドの平面性に起因するシストランスの化学交換が存在する。CF3 はカルバミドの N 原子から2結合分(-CONHCH2CF3)へだてているためその影響は顕著ではないが、低い温度ではピーク分裂やブロードニングが懸念されるため、推奨しない。筆者らは30℃の測定にて1本のピークとして観測されている(図2B)。
測定磁場
化学交換の速度が同じであっても化学交換している共鳴周波数(Hz)差が変化するため、測定磁場も重要である。低磁場では共鳴周波数差が小さくなり、速い交換状態(fast exchange)にあるスペクトルが得られやすい。高磁場では逆に共鳴周波数差が大きくなり、遅い交換状態(slow exchange)となりやすい。すなわち高温・低磁場では fast exchange よりに、低温・高磁場では slow exchange よりになる。
またグルタミンの CF3修飾における 19F-NMR 測定では、多核 NMR に特有の大きな化学シフト異方性による緩和の影響が顕著であり、高磁場化による分解能・感度向上は 1H-NMR ほど期待できない。
測定 pH
リジンの 13CH3 修飾においては、アミノ基のプロトン化状態およびそのプロトン交換速度がメチル基の化学シフトおよびピーク線形に影響をあたえる。pH を塩基性(pH >10)にするとアミノ基のプロトンが解離し、1H 化学シフトは 0.7 ppm ほど高磁場側に、13C 化学シフトは 1 ppm ほど低磁場側にシフトする(16)。ジメチルアミノ基の pKa はおよそ10であるため、もし pH 10 付近での測定が可能であるならば、残基ごとの pKa の違いにしたがって混み合ったピークを分散させることができる。また、pH をやや酸性より(pH <6)にするとアミノ基のプロトン交換速度が低下することに由来して、ピークが分裂またはブロードニングする場合がある(15–17)。
スペクトル変化の解析
分子間相互作用等にともなう構造変化をスペクトル変化から解析する場合、化学修飾によって導入した安定同位体標識部位はアミノ酸側鎖の末端であるため、化学シフト変化量自体は小さいことが多い。しかしながら、スペクトル全体からみた相対的な変化量は微細な化学環境の変化を鋭敏に反映する。リジンの 13CH3 修飾の場合では、1H 化学シフト変化量にして 0.01 ppm が、ひとつのしきい値となる(8)。13C 化学シフト変化も指標となりうるが相対的な変化量は 1H とくらべて小さく、また上記で述べた化学交換の影響をうけやすいため、1H 変化量のみを解析するほうが簡便である。グルタミンの CF3 修飾の場合、0.1 ppm 程度が有意な 19F 化学シフト変化量として期待される(9)。さらに、19F 核を含む多核に特有の広い化学シフト範囲および大きな化学シフト異方性によって、ピークブロードニングが引き起こされやすい傾向にある。
修飾反応の影響
実験の詳細1)で示したように、リジンの 13CH3 修飾では側鎖アミノ基の pKa が大きく変わらないため、中性 pH 付近ではプロトン化して正に帯電する。よって、酸性アミノ酸残基との塩橋といった静電的相互作用も保持されると考えられる。筆者らの研究では、メチル修飾した3種のタンパク質についての系統的な解析から、遅い化学交換によるピーク分裂と塩橋形成が強い相関を示し、逆に速い化学交換によって1本のピークとして観測される場合には露出した表面残基であることが示された(18)。またメチル修飾による立体構造変化は主鎖原子の RMSD にして 1 Å 以内であり、全体としてのフォールディングは保持される(8)。筆者らが NMR によって構造決定した FKBP12 のリジンメチル修飾体(PDB: 2ND5)では、非修飾体との主鎖 RMSD が 0.8 Å であった(18)。ただし、タンパク質の結晶化においてはリジンメチル修飾による格子間のパッキング効果によって結晶化能や分解能の向上がみられるように(19)、タンパク質の物性になにかしらの影響が起きることは考えうる。また、立体構造への影響という観点では表面残基よりも内部残基への修飾のほうが顕著である。そこで添加するホルムアルデヒドを10から 20 mM 程度に減らす、あるいは2, 3回に分けて試薬を添加するなど、修飾反応条件を最適化することによって内部残基への修飾をおさえることができる(8)。
トランスグルタミナーゼを用いたグルタミンの標識では、15N 標識の場合には化学構造が保持される。一方の 19F 標識における CF3 修飾では表面残基の疎水性が上がり、溶解度といった性状に影響を与え、場合によっては反応中に沈殿物が生じることがある。そのような場合は、2,2,2-Trifluoroethylamine hydrochloride の濃度や反応温度を下げるなどして、条件検討する。ただし沈殿物が生じた場合であっても、ある特定の残基が修飾された場合にのみ沈殿することがあるため、反応液の上清から他の残基がCF3修飾されたサンプルを得られることがある。
システインへの化学修飾
本稿ではリジンとグルタミンへの化学修飾について概説したが、部位選択的標識という観点ではシステインへの化学修飾がもっとも有力な手段となる。とりわけ CF3 修飾による 19F 標識は GPCR への応用が報告されるなど(20, 21)、化学修飾による安定同位体標識法において重要な位置をしめてきている。また安定同位体標識のほかにも常磁性タグ(スピンラベル)標識によって、NMR では常磁性緩和促進(PRE)や擬コンタクトシフト(PCS)といった常磁性効果による解析が、さらには電子スピン共鳴(EPR)への応用が可能となる。
実験の安全
リジンの還元的メチル化反応において Sodium cyanoborohydride を還元剤として用いる場合は、毒物及び劇物取締法に係るシアン化物であるため、手袋等で防護の上で取扱い、廃棄にあたっては法令にしたがう。
文献
- Wiesner, S. & Sprangers, R., Curr. Opin. Struct. Biol., 35, 60–67 (2015)
- Zhang, H. & van Ingen, H., Curr. Opin. Struct. Biol., 38, 75–82 (2016)
- Religa, T. L. et al., J. Am. Chem. Soc., 133, 9063–9068 (2011)
- Bokoch, M. P. et al., Nature, 463, 108–112 (2010)
- Jentoft, J. E. et al., J. Biol. Chem., 254, 4366–4370 (1979)
- Cabacungan, J. C. et al., Anal. Biochem., 124, 272–278 (1982)
- Rayment, I., Methods Enzymol., 276, 171–179 (1997)
- Hattori, Y. et al., J. Biomol. NMR, 55, 19–31 (2013)
- Hattori, Y. et al., J. Biomol. NMR, 68, 271–279 (2017)
- Shimba, N. et al., Anal. Biochem., 301, 123–127 (2002)
- Spolaore, B. et al., Biochemistry, 51, 8679–8689 (2012)
- Fontana, A. et al., Adv. Drug Deliver. Rev., 60, 130–28 (2008)
- Rachel, N. M. et al., Protein Sci., 26, 2268–2279 (2017)
- 服部 良一,児嶋 長次郎,生物物理,56,288–289 (2016)
- Gerken, T. A. et al., J. Biol. Chem., 257, 2894–2900 (1982)
- Zhang, M. & Vogel, H. J., J. Biol. Chem., 268, 22420–22428 (1993)
- Larda, S. T., et al., J. Biomol. NMR, 54, 199–209 (2012)
- Hattori, Y. et al., XXVIIth ICMRBS, Kyoto, Poster046-Mon (2016)
- Walter, T. S. et al., Structure, 14, 1617–1622 (2006)
- Liu, A. et al., Science, 335, 1106–1110 (2012)
- Manglik, A. et al., Cell, 161, 1101–1111 (2015)
