

概要
X 線によるタンパク質の立体構造解析が可能となって約70年、遺伝子工学・タンパク質工学の目覚ましい発展により、約14万件ものタンパク質の構造が報告され、生命機能の理解や分子標的薬の開発に利用されている。しかし、真核生物の主要な機能を担っている超分子複合体の解析は、試料調製の技術的な難しさから研究が難航している。本稿では分子量約 500 KDa、16サブユニットからなる転写メディエーターの研究を例として、超分子複合体の立体構造解析ストラタジーについて紹介したい。
イントロダクション
タンパク質の構造情報は生命科学研究だけでなく、医薬、食品、エネルギーなどの様々な産業の発展に寄与する重要な知見を与えてきた。しかし、医薬研究の標的であるヒトを含む真核生物のタンパク質の多くが、調製および解析が困難な巨大な複合体として機能しているため、需要に反して解析例が少ない。現在報告されているものは、RNA ポリメラーゼ II やリボソーム、プロテアソーム、シトクロム C オキシダーゼなど、天然宿主での発現量が多いタンパク質が主体となっている。
筆者らが立体構造解析を行った転写メディエーター(以下 MED)は、出芽酵母では25サブユニットから構成される約 1.4 MDa の超分子複合体であり、細胞での発現量が少ないことが構造解析を困難にしていた。これまで報告のあった精製法では、目的タンパク質のゲノムに直接 TAP タグを導入した、出芽酵母を数百リットルというスケールで培養し、内在性タンパク質を精製してくるために、プロテアーゼや多量の夾雑物を取り除くことが困難であった。
筆者らはこの問題を克服するために、MEDを構成する各サブユニットが比較的小さい分裂酵母由来のタンパク質を選択して、分裂酵母のコア・メディエーター(cMED)、16量体について大腸菌でのリコンビナントな発現系を樹立した(文献1)。本稿で紹介する 1) プラスミドの設計と複合体タンパク質の調製法、2) タンパク質の結晶化と回折実験、3) クロスリンク質量分析に用いるサンプルの調製法がほかの超分子複合体の立体構造解析にも応用できることを期待している。
装置・機器・試薬
- インキュベーター
- 5 L フラスコ
- 遠心機(Beckman)
- ソニケーター(Branson)
- AKTA purifier(GE Healthcare)
- HisTrap HP(GE Healthcare)
- HiTrap Q HP(GE Healthcare)
- Superose 6 10/300 GL(GE Healthcare)
- EasyXtal 15-Well Tool 結晶化プレート(Qiagen)
- Tantalum Cluster Derivatization Kit(Jena Bioscience)
- BS3-H12/D12(Thermo Fisher Scientific)
- Sequencing Grade Modified Trypsin(Promega)
- Ammonium bicarbonate BioUltra(Sigma)
実験手順
- プラスミドの設計と複合体タンパク質の調製
- タンパク質の結晶化と回折実験
- クロスリンク質量分析に用いるサンプルの調製法
実験の詳細
1. プラスミドの設計と複合体タンパク質の調製
16量体 cMED の発現系構築にあたり、それぞれの遺伝子は異なる薬剤(アンピシリン、カナマイシン、ストレプトマイシン)耐性遺伝子を持つ3つの大腸菌プラスミド(pET-21b、pET-28b、pETDuet-1)に、ポリシストロニックに組み込んだ(図1)。cMEDを構成するサブユニットは2つのモジュール(ヘッドとミドル)単位で機能することが知られていたため、同じモジュールに属するサブユニットは同じベクターに入るように設計し、共発現の前に各ベクターからのタンパク質の発現を確認した。通常、共発現に用いるプラスミド同士は、コピー数の問題から異なる複製起点を持つことが望ましいが、今回の2つのプラスミド(pET-21bとpET-28b)は同じ複製起点(pBR322)を有していたが問題なく発現した。pET-28b の代わりに複製起点の異なる pRSFDuet-1 についても発現の検討を行ったが、これは反って発現が見られず、複製起点の相性よりもベクターの相性の方が大事なケースもあると考えられた。各サブユニットの遺伝子の上流約 10 bp には、リボソーム結合配列(RBS)を配置し、mRNAの発現を助けるために T7 promoter 配列は約 1500 bp おきに挿入している。11 Kbp を超えるプラスミドでは大腸菌中のコピー数が減少してしまうという報告(文献2)もあるが、本研究で用いた 7~9 Kbp のプラスミドでは発現量に支障がないようだった。複合体中の各サブユニットの量比を合わせる工夫として、6xヒスチジンタグを最も発現量の少ない Med14 の N 末端に付加し、過分に発現したサブユニットは、精製中に外れるように設計した。
共発現に際しては、これら3つのベクターで大腸菌 BL21(DE3) RIL 株のケミカルコンピテントセルを形質転換した。手順としては、まずアンピシリン耐性とカナマイシン耐性のプラスミド(300 ng ずつ)で同時形質転換行った後(1日目)、2日目に得られたコロニーを10個程度集めて Transformation buffer(100 mM CaCl2, 10% glycerol)2 mL に懸濁し、30分間氷上で冷却することでケミカルコンピテントセル化した。菌体は遠心分離で回収し、50 μL 程度の Transformation buffer に再懸濁した後、3つ目のストレプトマイシン耐性のプラスミド 300 ng で2度目の形質転換とプレーティングを行った。
大腸菌の大量培養は LB 培地で行い、OD600 が0.7になるまで37℃で培養し、30分間氷上で菌体を冷却した後、終濃度 0.5 mMの IPTG でタンパク質の発現を誘導して、18℃で16時間培養した。回収した菌体は超音波破砕を行い、超遠心分離(15000 rpm、1時間)で得られた上清を Ni アフィニティークロマトグラフィー HisTrap HP、陰イオン交換クロマトグラフィー HiTrap Q HP、ゲルろ過クロマトグラフィー Superose 6 10/300 GL にかけることによってタンパク質を精製した。
精製産物の品質チェックは SDS-PAGE に展開した各バンドを質量分析にかけることによって行い、全てのサブユニットを含む複合体であることを確認した。最終的には 3 L の培養液から約 1 mg のタンパク質試料を得ることができた。
2. タンパク質の結晶化と回折実験
近年のクライオ電子顕微鏡解析の技術革新により、超分子複合体の解析が急速に進められているが、4 Å を下回るような中程度の分解能の場合、ヘテロなマルチサブユニットタンパク質の立体構造を決定するのは困難である。実際、MED の場合も多くの電子顕微鏡解析が試みられてきたが、おぼろげな概形しか決定できていなかったため、筆者らは X 線結晶構造解析を試みることにした。結晶化の初期スクリーニングをハンプトンリサーチ社やキアゲン社、モレキュラーディメンション社から販売されている各種キットで行った結果、得られた結晶からは 8 Å までの回折像しか得られなかったが、凍結前の結晶の加工と回折実験系の最適化を行うことで、分解能を改善することができた。
まず、結晶内の水分含有量をコントロールするデハイドレーション処理で結晶のパッキングを強固にして、分解能を 4 Å まで向上させた。デハイドレーションに用いる試薬と方法は、文献3と4を参考にした。ただし、多くの超分子複合体が4℃での処理で分解能の改善が見られていることから、本実験もデハイドレーション処理と結晶の凍結を4℃で行った。手順を以下に述べる(図2)。①20℃で作製した結晶と試薬、および器具類を予め前日から4℃に冷やしておく。②新しい結晶化プレートのリザーバーウェルにデハイドレーション試薬(30% PEG400 を含む結晶化溶液 500 μL)を加え、スクリューキャップには 7 μL のハーベスト溶液(1.2倍濃度の結晶化溶液)でドロップを用意した。③キャピラリーまたはピペットマンを使って結晶をスクリューキャップ上のドロップに移した。デハイドレーション中の結晶は空気にさらされるとダメージを受けてしまうため、ループでの結晶の移動は極力行わなかった。④結晶を壊さないように慎重にドロップ溶液を混合し、そのスクリューキャップでリザーバーウェルに蓋をして15分間インキュベートを行った。⑤ドロップから 7 μL の溶液を吸い取り、同量の5% PEG400 を含むハーベスト溶液を加えた。⑥ ④〜⑤の操作を10、15、20、25、30% PEG400 を含むハーベスト溶液を用いて(各濃度2回ずつ)順を追って繰り返した。⑦最終的に約30% PEG400 まで置換されたハーベスト溶液中で結晶を15分間インキュベートした後、その結晶をループですくって液体窒素で凍結保存した。
次にドイツ大型放射光施設 DESY のビームライン開発者である Thomas R. Schneider 博士と回折実験系を最適化し、cMED 結晶への X 線のダメージを軽減した。PETRA III シンクロトロンの P14 ビームラインの高輝度ビームを多段屈折レンズで集光させることで、並進性の高いX線(collimated ビーム)を発生させ、結晶サイズに合わせたビームを照射することで、分解能がさらに 0.6 Å 改善した。この方法では、ミラーを用いてビームサイズを小さく絞り、結晶上を移動させながら測定する従来の方法と比べて100倍以上の回折写真の枚数を測定することができた。
また、cMED には構造が全く未知の6つのサブユニット(Med1/ 9/ 4/ 14/ 10/ 19)も含まれていたため、既知の部分構造を用いた分子置換法のみでは位相を決定することができなかった。このため、筆者らは結晶に Ta6Br12 やセレノメチオニンを導入し、異常散乱法で位相を決定した。Ta6Br12 の導入は Native 結晶を Ta6Br12 溶液(2 mM の Ta6Br12 飽和水溶液 2 μL を 6 μL のハーベスト溶液と混合)に2時間浸すことで行った(図3)。cMED の場合、Ta6Br12 溶液に1時間浸した結晶に X 線を照射すると全く異常散乱のピークが得られず、3時間浸したものは結晶の見た目は変わらないものの、全く回折点が得られなかった。このため、結晶を Ta6Br12 溶液に浸す時間は標的タンパク質によって検討する必要があると考えられる。最終的に Ta6Br12 を導入した結晶からは、約 6 Å までの有効な異常散乱シグナルが得られ、セレノメチオニン置換体の結晶から得られた約 4 Å の異常散乱シグナルと既知の部分構造を用いて MR-SAD 法を行うことで、充分な位相情報が得られた。セレノメチオニン置換体の作成に関しては、蛋白質科学会アーカイブ「メチオニン要求株を使わないセレノメチオニン標識蛋白質の作り方」(文献5)を参照していただきたい。
位相情報が得られた後も、すべてのサブユニットを帰属させ、アミノ酸配列を決定するためには、Native 結晶に含まれるメチオニンやシステインの硫黄の位置情報を異常散乱によって測定する S-SAD 法のデータやセレノメチオニンを追加した変異体からの異常散乱シグナルが必要であった。
3. クロスリンク質量分析に用いるサンプルの調製法
結晶学的アプローチだけでは16サブユニットすべてを帰属するのは困難であったため、新しいアプローチとしてクロスリンク質量分析法(XL-MS)も用いた。XL-MS では、タンパク質中の相互作用を架橋剤で固定した後、プロテアーゼで消化し、得られた架橋ペプチドを質量分析法で同定することで、タンパク質の相互作用マップを作成することができる。cMED の実験では、リジン側鎖やタンパク質の N 末端の一級アミン基(-NH2)同士をスクシンイミド基を介して架橋する BS3 クロスリンカーを用いた。BS3 の Spacer Arm は 11.4 Å であることから、20〜30 Å のアミノ酸同士が架橋されることとなる。cMED の試料調製は、予検討(図4)を通じて下記の方法で行った。①終濃度 0.8 mg/mL のタンパク質 500 μg に対して終濃度 1.5 mM の BS3 を加えて、30℃、30分間架橋反応を行い、終濃度 50 mM の重炭酸アンモニウムを加えることで反応を停止した。なお、Tris や Glycine バッファーは架橋剤と反応してしまうため、実験には HEPES やリン酸バッファーを用いる必要がある。②反応液には過度に架橋されたタンパク質も混在しているため、ゲルろ過カラムで精製し、Native タンパク質と同じ位置に溶出される分画を回収した。③精製タンパク質はトリプシンプロテアーゼで切断し、約 100 μg のペプチドを複数回に分けて LC-MS/MS で分析した。ペプチドの分離には逆相カラム C18 を用いた。質量分析のために、架橋剤の BS3 は Spacer Arm の水素を重水素で置き換えた化合物も等量含まれる BS3-H12/D12 を用いている。BS3-H12/D12 で架橋されたペプチドは質量がずれた2つのピークとして現れ、分子量の近い未架橋ペプチドと容易に区別することができる。ペプチドの同定にはプログラム pLink(http://pfind.ict.ac.cn/software/pLink/)や ProteinProspector(http://prospector.ucsf.edu/prospector/mshome.htm)を用いた。④最後にプログラム xVis(https://xvis.genzentrum.lmu.de/CrossVisNoLogin.php)やタンパク質の構造ビューワー Chimera(https://www.cgl.ucsf.edu/chimera/)のプラグインである Xlink Analyzer(http://www.beck.embl.de/XlinkAnalyzer.html)を用いて、相互作用を図示化し構造のモデリングの指標とした(図5)。
構造解析の結果、Med1 は分子が結晶中でパッキングする際に欠損してしまったことが分かったが、酵母の生存に必要不可欠なサブユニットをすべて含む15サブユニット、約 450 KDa の cMED の結晶構造を 3.4 Å の分解能で決定することができた。
工夫とコツ
1. プラスミドの設計と複合体タンパク質の調製
cMED の粗精製物には多量のシャペロン・タンパク質が含まれるため、これを始めに取り除く必要があった。筆者らは、この問題を解決するために Ni アフィニティーカラムに cMED を吸着させた後、変性タンパク質(終濃度 2 mg/mL)とATP(終濃度 2 mM)を加えた洗浄バッファー(20 mM Tris-HCl_pH 8.0、500 mM 塩化カリウム、10%グリセロール、5 mM DTT)で担体を 10 CV 洗浄している。この過程でシャペロン・タンパク質は ATP により活性化され、cMED の代わりに変性タンパク質と結合して溶出される。なお、変性タンパク質の調製は形質転換を行っていない BL21 (DE3)RIL 株の破砕液から行い、超遠心した上清を20分間、60°Cで温め、再度超遠心分離して得られた上清を用いた。この方法で大腸菌の培養液 1 L から約 1 g の変性タンパク質を得ることができる。
解かれた cMED の立体構造を見ると、それぞれのサブユニットは単体では構造を支えられないような構造だったが、複合体として組み上がることによってはじめて、構造を保てるようなコンフォメーションをとっていた。このようにフォールディングが難しい性質のために、cMED にはシャペロン・タンパク質が結合していたと考えられる。
2. タンパク質の結晶化と回折実験
セレノメチオニン変異を入れる cMED のアミノ酸残基は、2次構造予測プログラム PSIPRED(http://bioinf.cs.ucl.ac.uk/psipred/)で構造をとると予想される領域の中で、生物種間の保存性が高いロイシンまたはイソロイシンを選定し、総数29ヶ所を置換した3つの変異体を作成した。
3. クロスリンク質量分析に用いるサンプルの調製法
XL-MS を行うにあたり、ターゲットとなるタンパク質にあった架橋剤とプロテアーゼの選定が必要となる。解析したいタンパク質領域にリジンがない場合は、当然ながら BS3 では架橋されない。また、もし標的タンパク質にリジンもアルギニンも少ない場合は、トリプシンで切断される間隔が広くなってしまい、質量分析で正しく検出されない可能性もある。逆に、リジンやアルギニンが連続して存在している場合には、トリプシン消化の際にペプチドが細かくなり過ぎてしまい、ペプチドを同定できない可能性もある。XL-MS を行う前には、目的タンパク質の配列を改めてチェックしていただきたい。
文献
- Nozawa, K., Schneider, TR. & Cramer, P., Nature, 545, 248–251 (2017)
- Smith, MA. & Bidochka, MJ., Can. J. Microbiol., 44, 351–355 (1998)
- Deng, X., Davidson, WS. & Thompson, TB., Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 68, 105–110 (2012)
- Heras, B. & Martin, JL., Acta. Crystallogr. D Biol. Crystallogr., 61, 1173–1180 (2005)
- 中村努 & 石川一彦, 蛋白質科学会アーカイブ, 1, e045 (2008)
変更履歴
| 変更日 | 変更内容 | 変更前の PDF |
|---|---|---|
| 2019/02/20 | 「装置・機器・試薬」と「実験の詳細」に記載の「Superose 6 10/600 GL」を「Superose 6 10/300 GL」に訂正、「実験の詳細」に記載の共発現用のトランスフォーメーションに用いるプラスミドの量「3 ng」を「300 ng」に訂正 | 変更前の PDF |
-
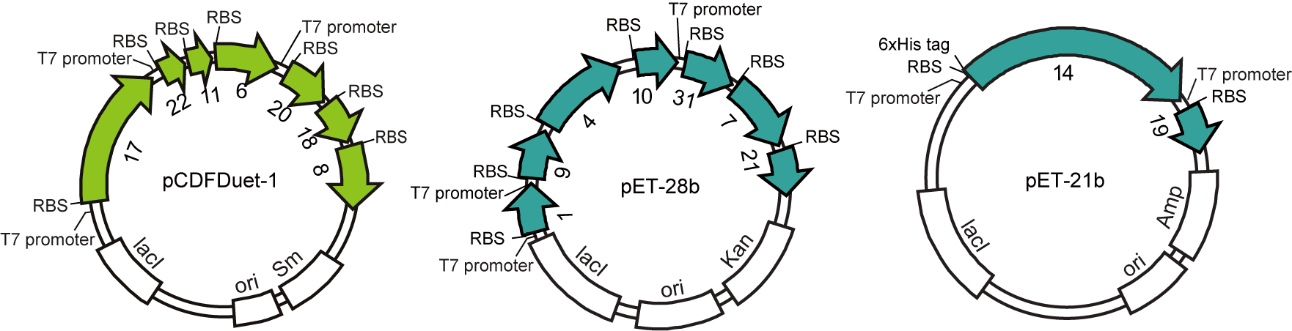
図1: cMED 発現プラスミドの模式図。黄緑はヘッド・モジュール、青緑はミドル・モジュールに属するサブユニットの遺伝子を示す。 -

図2: デハイドレーション操作の模式図。 -

図3: 結晶への Ta6Br12 の導入。上段は Ta6Br12 の構造式、下段は Ta6Br12 溶液に2時間浸した cMED の結晶写真。 -
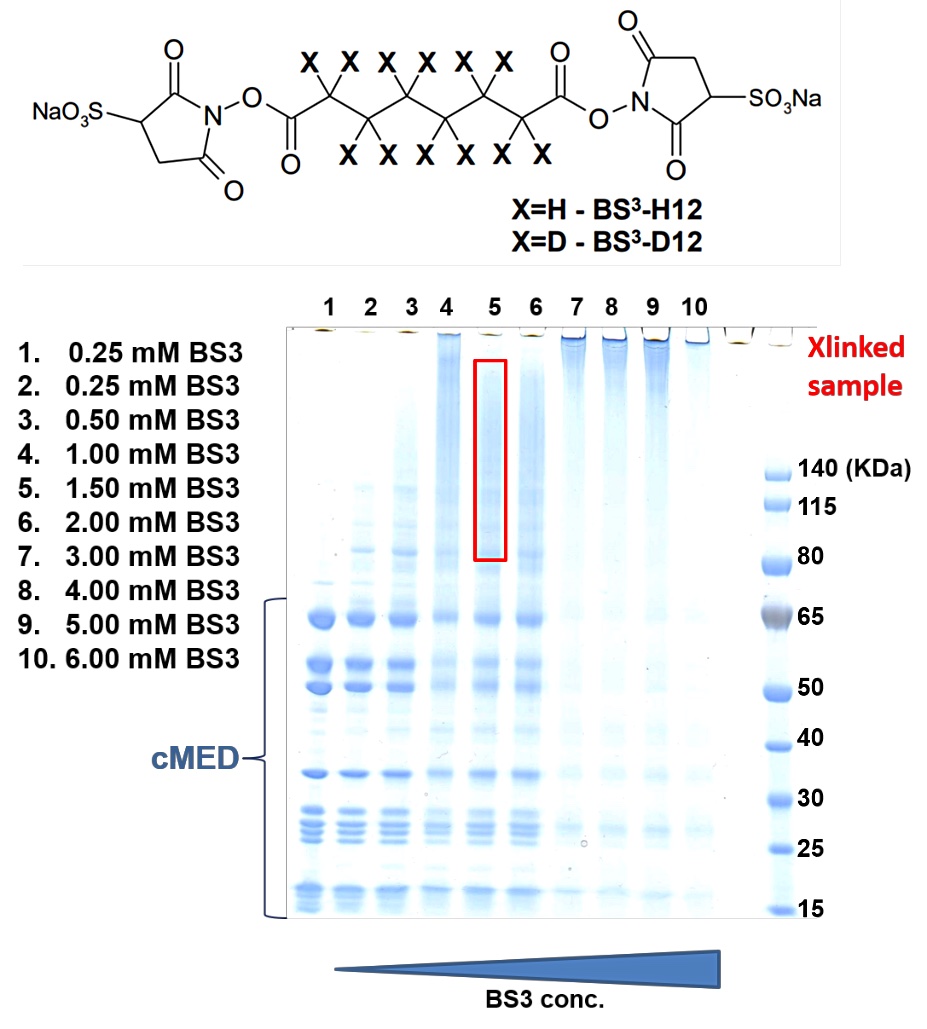
図4: 架橋剤 BS3 の検討実験。上段は、実験に用いた架橋剤 BS3 の構造式、下段は、検討実験で得られた SDS-PAGE を示す。反応条件は30℃、30分間に固定して、BS3 の濃度検討を行った。7レーン目以降は、過度に架橋されたタンパク質がアグリゲーションを起こしている。 -
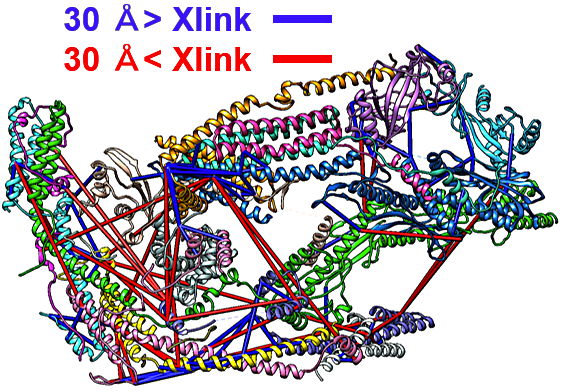
図5: XL-MS 解析から得られた cMED タンパク質の相互作用図。Xlink Analyzer を用いて図示化している。
概要
X 線によるタンパク質の立体構造解析が可能となって約70年、遺伝子工学・タンパク質工学の目覚ましい発展により、約14万件ものタンパク質の構造が報告され、生命機能の理解や分子標的薬の開発に利用されている。しかし、真核生物の主要な機能を担っている超分子複合体の解析は、試料調製の技術的な難しさから研究が難航している。本稿では分子量約 500 KDa、16サブユニットからなる転写メディエーターの研究を例として、超分子複合体の立体構造解析ストラタジーについて紹介したい。
イントロダクション
タンパク質の構造情報は生命科学研究だけでなく、医薬、食品、エネルギーなどの様々な産業の発展に寄与する重要な知見を与えてきた。しかし、医薬研究の標的であるヒトを含む真核生物のタンパク質の多くが、調製および解析が困難な巨大な複合体として機能しているため、需要に反して解析例が少ない。現在報告されているものは、RNA ポリメラーゼ II やリボソーム、プロテアソーム、シトクロム C オキシダーゼなど、天然宿主での発現量が多いタンパク質が主体となっている。
筆者らが立体構造解析を行った転写メディエーター(以下 MED)は、出芽酵母では25サブユニットから構成される約 1.4 MDa の超分子複合体であり、細胞での発現量が少ないことが構造解析を困難にしていた。これまで報告のあった精製法では、目的タンパク質のゲノムに直接 TAP タグを導入した、出芽酵母を数百リットルというスケールで培養し、内在性タンパク質を精製してくるために、プロテアーゼや多量の夾雑物を取り除くことが困難であった。
筆者らはこの問題を克服するために、MEDを構成する各サブユニットが比較的小さい分裂酵母由来のタンパク質を選択して、分裂酵母のコア・メディエーター(cMED)、16量体について大腸菌でのリコンビナントな発現系を樹立した(文献1)。本稿で紹介する 1) プラスミドの設計と複合体タンパク質の調製法、2) タンパク質の結晶化と回折実験、3) クロスリンク質量分析に用いるサンプルの調製法がほかの超分子複合体の立体構造解析にも応用できることを期待している。
装置・機器・試薬
- インキュベーター
- 5 L フラスコ
- 遠心機(Beckman)
- ソニケーター(Branson)
- AKTA purifier(GE Healthcare)
- HisTrap HP(GE Healthcare)
- HiTrap Q HP(GE Healthcare)
- Superose 6 10/300 GL(GE Healthcare)
- EasyXtal 15-Well Tool 結晶化プレート(Qiagen)
- Tantalum Cluster Derivatization Kit(Jena Bioscience)
- BS3-H12/D12(Thermo Fisher Scientific)
- Sequencing Grade Modified Trypsin(Promega)
- Ammonium bicarbonate BioUltra(Sigma)
実験手順
- プラスミドの設計と複合体タンパク質の調製
- タンパク質の結晶化と回折実験
- クロスリンク質量分析に用いるサンプルの調製法
実験の詳細
1. プラスミドの設計と複合体タンパク質の調製
16量体 cMED の発現系構築にあたり、それぞれの遺伝子は異なる薬剤(アンピシリン、カナマイシン、ストレプトマイシン)耐性遺伝子を持つ3つの大腸菌プラスミド(pET-21b、pET-28b、pETDuet-1)に、ポリシストロニックに組み込んだ(図1)。cMEDを構成するサブユニットは2つのモジュール(ヘッドとミドル)単位で機能することが知られていたため、同じモジュールに属するサブユニットは同じベクターに入るように設計し、共発現の前に各ベクターからのタンパク質の発現を確認した。通常、共発現に用いるプラスミド同士は、コピー数の問題から異なる複製起点を持つことが望ましいが、今回の2つのプラスミド(pET-21bとpET-28b)は同じ複製起点(pBR322)を有していたが問題なく発現した。pET-28b の代わりに複製起点の異なる pRSFDuet-1 についても発現の検討を行ったが、これは反って発現が見られず、複製起点の相性よりもベクターの相性の方が大事なケースもあると考えられた。各サブユニットの遺伝子の上流約 10 bp には、リボソーム結合配列(RBS)を配置し、mRNAの発現を助けるために T7 promoter 配列は約 1500 bp おきに挿入している。11 Kbp を超えるプラスミドでは大腸菌中のコピー数が減少してしまうという報告(文献2)もあるが、本研究で用いた 7~9 Kbp のプラスミドでは発現量に支障がないようだった。複合体中の各サブユニットの量比を合わせる工夫として、6xヒスチジンタグを最も発現量の少ない Med14 の N 末端に付加し、過分に発現したサブユニットは、精製中に外れるように設計した。
共発現に際しては、これら3つのベクターで大腸菌 BL21(DE3) RIL 株のケミカルコンピテントセルを形質転換した。手順としては、まずアンピシリン耐性とカナマイシン耐性のプラスミド(300 ng ずつ)で同時形質転換行った後(1日目)、2日目に得られたコロニーを10個程度集めて Transformation buffer(100 mM CaCl2, 10% glycerol)2 mL に懸濁し、30分間氷上で冷却することでケミカルコンピテントセル化した。菌体は遠心分離で回収し、50 μL 程度の Transformation buffer に再懸濁した後、3つ目のストレプトマイシン耐性のプラスミド 300 ng で2度目の形質転換とプレーティングを行った。
大腸菌の大量培養は LB 培地で行い、OD600 が0.7になるまで37℃で培養し、30分間氷上で菌体を冷却した後、終濃度 0.5 mMの IPTG でタンパク質の発現を誘導して、18℃で16時間培養した。回収した菌体は超音波破砕を行い、超遠心分離(15000 rpm、1時間)で得られた上清を Ni アフィニティークロマトグラフィー HisTrap HP、陰イオン交換クロマトグラフィー HiTrap Q HP、ゲルろ過クロマトグラフィー Superose 6 10/300 GL にかけることによってタンパク質を精製した。
精製産物の品質チェックは SDS-PAGE に展開した各バンドを質量分析にかけることによって行い、全てのサブユニットを含む複合体であることを確認した。最終的には 3 L の培養液から約 1 mg のタンパク質試料を得ることができた。
2. タンパク質の結晶化と回折実験
近年のクライオ電子顕微鏡解析の技術革新により、超分子複合体の解析が急速に進められているが、4 Å を下回るような中程度の分解能の場合、ヘテロなマルチサブユニットタンパク質の立体構造を決定するのは困難である。実際、MED の場合も多くの電子顕微鏡解析が試みられてきたが、おぼろげな概形しか決定できていなかったため、筆者らは X 線結晶構造解析を試みることにした。結晶化の初期スクリーニングをハンプトンリサーチ社やキアゲン社、モレキュラーディメンション社から販売されている各種キットで行った結果、得られた結晶からは 8 Å までの回折像しか得られなかったが、凍結前の結晶の加工と回折実験系の最適化を行うことで、分解能を改善することができた。
まず、結晶内の水分含有量をコントロールするデハイドレーション処理で結晶のパッキングを強固にして、分解能を 4 Å まで向上させた。デハイドレーションに用いる試薬と方法は、文献3と4を参考にした。ただし、多くの超分子複合体が4℃での処理で分解能の改善が見られていることから、本実験もデハイドレーション処理と結晶の凍結を4℃で行った。手順を以下に述べる(図2)。①20℃で作製した結晶と試薬、および器具類を予め前日から4℃に冷やしておく。②新しい結晶化プレートのリザーバーウェルにデハイドレーション試薬(30% PEG400 を含む結晶化溶液 500 μL)を加え、スクリューキャップには 7 μL のハーベスト溶液(1.2倍濃度の結晶化溶液)でドロップを用意した。③キャピラリーまたはピペットマンを使って結晶をスクリューキャップ上のドロップに移した。デハイドレーション中の結晶は空気にさらされるとダメージを受けてしまうため、ループでの結晶の移動は極力行わなかった。④結晶を壊さないように慎重にドロップ溶液を混合し、そのスクリューキャップでリザーバーウェルに蓋をして15分間インキュベートを行った。⑤ドロップから 7 μL の溶液を吸い取り、同量の5% PEG400 を含むハーベスト溶液を加えた。⑥ ④〜⑤の操作を10、15、20、25、30% PEG400 を含むハーベスト溶液を用いて(各濃度2回ずつ)順を追って繰り返した。⑦最終的に約30% PEG400 まで置換されたハーベスト溶液中で結晶を15分間インキュベートした後、その結晶をループですくって液体窒素で凍結保存した。
次にドイツ大型放射光施設 DESY のビームライン開発者である Thomas R. Schneider 博士と回折実験系を最適化し、cMED 結晶への X 線のダメージを軽減した。PETRA III シンクロトロンの P14 ビームラインの高輝度ビームを多段屈折レンズで集光させることで、並進性の高いX線(collimated ビーム)を発生させ、結晶サイズに合わせたビームを照射することで、分解能がさらに 0.6 Å 改善した。この方法では、ミラーを用いてビームサイズを小さく絞り、結晶上を移動させながら測定する従来の方法と比べて100倍以上の回折写真の枚数を測定することができた。
また、cMED には構造が全く未知の6つのサブユニット(Med1/ 9/ 4/ 14/ 10/ 19)も含まれていたため、既知の部分構造を用いた分子置換法のみでは位相を決定することができなかった。このため、筆者らは結晶に Ta6Br12 やセレノメチオニンを導入し、異常散乱法で位相を決定した。Ta6Br12 の導入は Native 結晶を Ta6Br12 溶液(2 mM の Ta6Br12 飽和水溶液 2 μL を 6 μL のハーベスト溶液と混合)に2時間浸すことで行った(図3)。cMED の場合、Ta6Br12 溶液に1時間浸した結晶に X 線を照射すると全く異常散乱のピークが得られず、3時間浸したものは結晶の見た目は変わらないものの、全く回折点が得られなかった。このため、結晶を Ta6Br12 溶液に浸す時間は標的タンパク質によって検討する必要があると考えられる。最終的に Ta6Br12 を導入した結晶からは、約 6 Å までの有効な異常散乱シグナルが得られ、セレノメチオニン置換体の結晶から得られた約 4 Å の異常散乱シグナルと既知の部分構造を用いて MR-SAD 法を行うことで、充分な位相情報が得られた。セレノメチオニン置換体の作成に関しては、蛋白質科学会アーカイブ「メチオニン要求株を使わないセレノメチオニン標識蛋白質の作り方」(文献5)を参照していただきたい。
位相情報が得られた後も、すべてのサブユニットを帰属させ、アミノ酸配列を決定するためには、Native 結晶に含まれるメチオニンやシステインの硫黄の位置情報を異常散乱によって測定する S-SAD 法のデータやセレノメチオニンを追加した変異体からの異常散乱シグナルが必要であった。
3. クロスリンク質量分析に用いるサンプルの調製法
結晶学的アプローチだけでは16サブユニットすべてを帰属するのは困難であったため、新しいアプローチとしてクロスリンク質量分析法(XL-MS)も用いた。XL-MS では、タンパク質中の相互作用を架橋剤で固定した後、プロテアーゼで消化し、得られた架橋ペプチドを質量分析法で同定することで、タンパク質の相互作用マップを作成することができる。cMED の実験では、リジン側鎖やタンパク質の N 末端の一級アミン基(-NH2)同士をスクシンイミド基を介して架橋する BS3 クロスリンカーを用いた。BS3 の Spacer Arm は 11.4 Å であることから、20〜30 Å のアミノ酸同士が架橋されることとなる。cMED の試料調製は、予検討(図4)を通じて下記の方法で行った。①終濃度 0.8 mg/mL のタンパク質 500 μg に対して終濃度 1.5 mM の BS3 を加えて、30℃、30分間架橋反応を行い、終濃度 50 mM の重炭酸アンモニウムを加えることで反応を停止した。なお、Tris や Glycine バッファーは架橋剤と反応してしまうため、実験には HEPES やリン酸バッファーを用いる必要がある。②反応液には過度に架橋されたタンパク質も混在しているため、ゲルろ過カラムで精製し、Native タンパク質と同じ位置に溶出される分画を回収した。③精製タンパク質はトリプシンプロテアーゼで切断し、約 100 μg のペプチドを複数回に分けて LC-MS/MS で分析した。ペプチドの分離には逆相カラム C18 を用いた。質量分析のために、架橋剤の BS3 は Spacer Arm の水素を重水素で置き換えた化合物も等量含まれる BS3-H12/D12 を用いている。BS3-H12/D12 で架橋されたペプチドは質量がずれた2つのピークとして現れ、分子量の近い未架橋ペプチドと容易に区別することができる。ペプチドの同定にはプログラム pLink(http://pfind.ict.ac.cn/software/pLink/)や ProteinProspector(http://prospector.ucsf.edu/prospector/mshome.htm)を用いた。④最後にプログラム xVis(https://xvis.genzentrum.lmu.de/CrossVisNoLogin.php)やタンパク質の構造ビューワー Chimera(https://www.cgl.ucsf.edu/chimera/)のプラグインである Xlink Analyzer(http://www.beck.embl.de/XlinkAnalyzer.html)を用いて、相互作用を図示化し構造のモデリングの指標とした(図5)。
構造解析の結果、Med1 は分子が結晶中でパッキングする際に欠損してしまったことが分かったが、酵母の生存に必要不可欠なサブユニットをすべて含む15サブユニット、約 450 KDa の cMED の結晶構造を 3.4 Å の分解能で決定することができた。
工夫とコツ
1. プラスミドの設計と複合体タンパク質の調製
cMED の粗精製物には多量のシャペロン・タンパク質が含まれるため、これを始めに取り除く必要があった。筆者らは、この問題を解決するために Ni アフィニティーカラムに cMED を吸着させた後、変性タンパク質(終濃度 2 mg/mL)とATP(終濃度 2 mM)を加えた洗浄バッファー(20 mM Tris-HCl_pH 8.0、500 mM 塩化カリウム、10%グリセロール、5 mM DTT)で担体を 10 CV 洗浄している。この過程でシャペロン・タンパク質は ATP により活性化され、cMED の代わりに変性タンパク質と結合して溶出される。なお、変性タンパク質の調製は形質転換を行っていない BL21 (DE3)RIL 株の破砕液から行い、超遠心した上清を20分間、60°Cで温め、再度超遠心分離して得られた上清を用いた。この方法で大腸菌の培養液 1 L から約 1 g の変性タンパク質を得ることができる。
解かれた cMED の立体構造を見ると、それぞれのサブユニットは単体では構造を支えられないような構造だったが、複合体として組み上がることによってはじめて、構造を保てるようなコンフォメーションをとっていた。このようにフォールディングが難しい性質のために、cMED にはシャペロン・タンパク質が結合していたと考えられる。
2. タンパク質の結晶化と回折実験
セレノメチオニン変異を入れる cMED のアミノ酸残基は、2次構造予測プログラム PSIPRED(http://bioinf.cs.ucl.ac.uk/psipred/)で構造をとると予想される領域の中で、生物種間の保存性が高いロイシンまたはイソロイシンを選定し、総数29ヶ所を置換した3つの変異体を作成した。
3. クロスリンク質量分析に用いるサンプルの調製法
XL-MS を行うにあたり、ターゲットとなるタンパク質にあった架橋剤とプロテアーゼの選定が必要となる。解析したいタンパク質領域にリジンがない場合は、当然ながら BS3 では架橋されない。また、もし標的タンパク質にリジンもアルギニンも少ない場合は、トリプシンで切断される間隔が広くなってしまい、質量分析で正しく検出されない可能性もある。逆に、リジンやアルギニンが連続して存在している場合には、トリプシン消化の際にペプチドが細かくなり過ぎてしまい、ペプチドを同定できない可能性もある。XL-MS を行う前には、目的タンパク質の配列を改めてチェックしていただきたい。
文献
- Nozawa, K., Schneider, TR. & Cramer, P., Nature, 545, 248–251 (2017)
- Smith, MA. & Bidochka, MJ., Can. J. Microbiol., 44, 351–355 (1998)
- Deng, X., Davidson, WS. & Thompson, TB., Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 68, 105–110 (2012)
- Heras, B. & Martin, JL., Acta. Crystallogr. D Biol. Crystallogr., 61, 1173–1180 (2005)
- 中村努 & 石川一彦, 蛋白質科学会アーカイブ, 1, e045 (2008)
変更履歴
| 変更日 | 変更内容 | 変更前の PDF |
|---|---|---|
| 2019/02/20 | 「装置・機器・試薬」と「実験の詳細」に記載の「Superose 6 10/600 GL」を「Superose 6 10/300 GL」に訂正、「実験の詳細」に記載の共発現用のトランスフォーメーションに用いるプラスミドの量「3 ng」を「300 ng」に訂正 | 変更前の PDF |
