

概要
二成分毒素は種々の細菌が産生する、酵素活性を持つ A 成分と膜結合性の B 成分からなるタンパク質毒素である。B 成分は標的細胞膜上で環状の可溶性オリゴマー(プレ膜孔)を形成すると、これに A 成分が結合し複合体となり、エンドソームに取り込まれる。エンドソーム内の pH が低下するとプレ膜孔は膜貫通型のβバレルを形成して膜孔に構造変化する。A 成分はこの中を通過して細胞内に侵入すると考えられているが、その透過機構の詳細は明らかにされていなかった。我々は A 成分と B 成分膜孔複合体の構造決定をクライオ電子顕微鏡を用いて行った (1)。ここでは、二成分毒素の透過機構の一端を明らかにした、イオタ毒素の膜透過装置である Ib 膜孔、さらに酵素成分 Ia が結合した Ia–Ib 膜孔複合体の調製に注目して方法を記す。構造から得られた知見については文献 (1) をご参考下さればと思う。
目的・イントロダクション等
二成分毒素は種々の細菌が産生する、酵素活性を持つ A 成分と膜結合性の B 成分からなるタンパク質毒素である (2)。B 成分は可溶性モノマーとして産生され、標的細胞膜上の受容体に結合する。宿主が有するプロテアーゼによって N 末端の約 20 kDa の Pro 配列が切断されることで活性化され、可溶性オリゴマー(プレ膜孔)を形成する (3)(4)。A 成分はプレ膜孔に結合し、この複合体はエンドソームに取り込まれる。エンドソーム内の pH が低下するとプレ膜孔は膜貫通型のβバレルを有する膜孔に構造変化する (5)。A 成分はエンドソーム内部の低 pH から細胞質側の中性 pH にかけてのプロトン勾配を利用して透過する。細胞質側に透過した A 成分は酵素活性を発揮し、標的細胞に重篤な影響を及ぼす。
炭疽菌(Bacillus anthracis)が産生する二成分毒素である炭疽菌毒素では、クライオ電子顕微鏡による単粒子解析で膜孔の構造が明らかにされた (6)。この報告により種々の細菌で高度に保存される B 成分膜孔は、A 成分が通過すると考えられる経路に直径わずか 6 Å の狭窄部位φクランプを有することが明らかにされた。A 成分がφクランプを通過するためにはこの狭窄部位が拡張する、あるいは A 成分が構造を失う必要があるがその詳細は不明である。
そこで我々は膜孔の構造が未知であった、膜結合成分 Ib とアクチンを標的とする ADP リボシル化毒素 Ia から構成されるイオタ毒素 (7)(8) の、Ib 膜孔および、Ia–Ib 膜孔複合体のサンプル調製を行い、クライオ電子顕微鏡を用いた単粒子解析によってそれぞれの構造を 3.0 Å を切る高分解で明らかにした(図1)(1)。すでに報告されていたIaの単体の結晶構造と本研究で明らかにした複合体の構造の比較から、Ia は Ib 膜孔への結合によってN末端のαヘリックスがアンフォールドすることがわかった。さらに構造を失った領域がφクランプまで続いているかのようなマップを観察することもできた。
構造解析によって得られたこれらの知見は二成分毒素を標的にした化合物開発の構造基盤となるだけでなく、膜を介したタンパク質の透過に対して重要な知見をもたらした。二成分毒素の研究は任意の分子を細胞内に届けるアプリケーションとしての応用などにも発展することが期待できる。ここではイオタ毒素の構造解析において最も難関であった Ib 膜孔の調製方法を記す。さらに Ia との複合体調製から単粒子構造解析に向けたグリッド作製についても触れる。このプロトコールを今後の二成分毒素などの膜タンパク質調製への参考にして下さればと思う。
装置・器具・試薬
培養に必要なもの
- 大腸菌:Escherichia Coli origami B (DE3)
- 大腸菌:Escherichia Coli BL21 (DE3)
- ベクター:pGEX4T-1
- ベクター:pET-21b
- アンピシリン
- テトラサイクリン
- カナマイシン
- LB 固体培地
- LB 液体培地
- Super Broth 液体培地
- 15 mL チューブ用恒温震盪培養器
- 5 L フラスコ用震盪培養器
- 濁度計
- IPTG(Isopropyl-β-D-thiogalactopyranoside)
精製・オリゴマー化処理に必要なもの
- cOmplete(Protease inhibitor cocktail)
- 金属カップ
- ソニケーター
- Glutathione Sepharose 4B resin(GE healthcare)
- DTT(dithiothreitol)
- 還元型グルタチオン
- ナノドロップ
- 遠心用チューブ30K、50Kおよび100K(Amicon)
- Chymotrypsin(SIGMA)
- PMSF(Wako)
- LMNG(Anatrace)
- P40ST(スイングローター)
- 13PA チューブクミ(himac)
- 注射針(TERUMO)
- PD-10(GE healthcare)
- イミダゾール
- Ni-NTA 樹脂
- Hi-Trap Q HP 5ml(GE healthcare)
実験手順
- Ibの膜孔形成
- クローニングおよび培養
- Ib 膜孔の精製
- オリゴマー形成
- Ib 膜孔の精製
- 密度勾配遠心
- フラクション分け
- Buffer 交換
- Ia の調製
- クローニングおよび培養
- Ia の精製
- 複合体の形成およびグリッド作製
- 複合体形成
- グリッド作製
実験の詳細
Ib の膜孔形成
Ib は大腸菌によって可溶性のモノマーとして発現させ、GST タグを使用して精製する。精製した Ib モノマーは Chymotrypsin 処理によって活性化させ、7量体のオリゴマーである膜孔に構造変化させる。Ib 膜孔は膜貫通型のタンパク質であるため、可溶化をすることが求められる。電子顕微鏡解析に向けた膜タンパク質の可溶化には細胞の脂質環境を模倣したリポソームや、脂質二重膜が MSP(membrane scaffold protein)というタンパク質で囲まれて円盤状の形をしたナノディスク (9)(10) への再構成、あるいは界面活性剤ミセルで疎水部分を覆うことで可溶化する方法などがよく使われる。本プロトコルの Ib 膜孔の可溶化には LMNG を使用して可溶化を行った。LMNG は臨界ミセル濃度 Critical micelle concentration(CMC)=0.001% (w/v) 以下においても、膜タンパク質を可溶化することができるという特徴を持つ。このため適切な低濃度で使用すれば電子顕微鏡画像に不必要なミセルが現れにくい。
Ib 膜孔においては実際に極めて低い LMNG 濃度でも十分に可溶化されたサンプルを調製できることがわかった。電子顕微鏡試料の LMNG 濃度の検討の末、本プロトコルでは0.003% (w/v) の低濃度 LMNG を含む条件で調製した例を示す。
1. クローニングおよび培養
Ib 遺伝子(Uniplot ID:Q46221)が導入された pGEX4T-1 ベクターを用意し、これを大腸菌 E. coli origami にヒートショック法で形質転換させる。これをアンピシリン(50 μg/ml)、テトラサイクリン(12.5 μg/ml)およびカナマイシン(15 μg/ml)を含むLB固体培地に塗布し、37℃で一晩培養する。翌日、固体培地上に形成されたコロニーを用いて、アンピシリン(50 μg/ml)、テトラサイクリン(12.5 μg/ml)およびカナマイシン(15 μg/ml)を含む 3 ml の LB 液体培地に移し、37℃の温浴槽で一晩震盪培養する。培養された菌体液とあらかじめオートクレーブされた50% (v/v) グリセロール溶液と混合し、これをグリセロールストックとして−80℃で冷凍保存する。
少量のグリセロールストックをアンピシリン(50 μg/ml)、テトラサイクリン(12.5 μg/ml)およびカナマイシン(15 μg/ml)を含む 3 ml の Super Broth 液体培地に加え、37℃の温浴槽で一晩震盪培養する。翌日、菌体液全量をアンピシリン(50 μg/ml)、テトラサイクリン(12.5 μg/ml)およびカナマイシン(15 μg/ml)を含む 1 L の Super Broth 液体培地に加え、37℃で震盪培養する。Super Broth 液体培地をベースラインとして、培養液の濁度(OD600)=0.6(AU)となったところで培養を一度止め、培養液の入ったフラスコを氷水で冷やす。十分に冷却された培養液に終濃度 1 mM となるように IPTG を加え、23℃で一晩震盪培養する。培養液を4℃、12,092 ×g で5分間遠心分離する。沈降した菌体を 50 ml チューブにスパーテルなどで移し替え、−80℃で冷凍保存する。
2. Ib 膜孔の精製
あらかじめ下記の Buffer を用意する。
- Buffer A: 20 mM Tris pH 8.0,150 mM \(\ce{NaCl}\),2 mM \(\ce{CaCl2}\),5 mM DTT
- Buffer B: 20 mM Tris pH 8.0,150 mM \(\ce{NaCl}\),2 mM \(\ce{CaCl2}\),10 mM 還元型グルタチオン
- Buffer C: 20 mM Tris pH 8.0,50 ml \(\ce{NaCl}\),2.5 mM \(\ce{CaCl2}\)
冷凍保存した菌体を室温で融解し一錠の cOmplete を溶解させた 120 ml の Buffer A に懸濁する。これを金属製のカップに移し、氷水上でソニケーションによって大腸菌を破砕する。破砕菌体液を超遠心用のチューブに移し替え、4℃、180,000 ×g で40分間、超遠心にかける。上清をあらかじめ Buffer A に置換された体積 5 ml の Glutathione Sepharose 4B resin 樹脂と4℃で1時間転倒混和する。樹脂からフロースルー回収後、Buffer A を用いて1フラクション 5 ml の wash を行う。ナノドロップによって wash 画分の濃度(A280nm)を計測し、これが 0.1 mg/ml を下回るまで wash を繰り返す。次に Buffer B を用いて1フラクション 1 ml の Elution を行う。溶出されたタンパク質の濃度(A280nm)がピークを超えた後に 0.1 mg/ml を下回るまで Elution を繰り返す。得られた Elution 画分を集め、Amicon 50K によって Buffer C ヘの交換と濃縮を行う。濃縮されたサンプルは 500 μl 程度になるように分注し、液体窒素による瞬間凍結の後、−80℃で冷凍保存する。
3. オリゴマー形成
2. で得られた GST タグ–Pro 配列–Ib に対して質量比で1000分の1量の Chymotrypsin を加える(図2.a)。室温(25℃程度)で1時間インキュベートし、Pro 配列–Ib 間を Chymotrypsin によって切断させる。反応液に終濃度 1 mM PMSF、10% (v/v) エタノールおよび0.03% (w/v) LMNG となるように加え、37℃で1時間インキュベートする(図2.b)。
Ib 膜孔の精製
7量体のオリゴマー Ib 膜孔は分子量 500 kDa を超える膜タンパク質である。このような巨大な膜タンパク質はゲル濾過のような手法で精製することが多い。本プロトコルではゲル濾過と同様に分子量依存的なタンパク質の分離方法である、密度勾配遠心という手法で Ib 膜孔を精製した。ゲル濾過とは異なりカラムを必要とせず、一連の実験を超遠心用のチューブの中で行う。そのため凝集したタンパク質によるカラム詰まりのリスクが無い。当研究初期は Ib 膜孔が LMNG 存在下においても、十分に可溶化された状態であるのか確信を持てなかったため、この手法を用いることにした。可溶化した膜タンパク質溶液から遊離ミセル除去する方法として、GraDeR という手法が報告されている (11)。GraDeR も本プロトコルと同じく LMNG 存在下での密度勾配遠心を行う。GraDeR においては緩やかに遊離ミセルを取り除くためにサンプルをアプライするチューブ上部側から反対の底部にかけて LMNG 濃度勾配を生じさせるが、今回紹介するプロトコルでは遠心チューブ全体で LMNG 濃度は均一である。
オリゴマー形成時には Ib 膜孔の可溶化を目的に0.03% (w/v) LMNG を使用している。しかしながらこの LMNG 濃度を保ったまま精製を行う必要はなく、密度勾配遠心以降に LMNG 濃度を0.003% (w/v) に落としている。不要に高濃度の LMNG を使用すると電子顕微鏡画像にミセルと思われる像が現れてしまう(実際に0.01%–0.005% (w/v) LMNG を使用するとミセルらしき像が大量に現れてしまった)。ミセルの出現は LMNG を含まない buffer と希釈することで軽減することが可能であるが、画像あたりの粒子数も減らしてしまうことになるため、あらかじめ LMNG 濃度を低くしておくに越したことはない。
1. 密度勾配遠心
下記の Buffer を用意する。
- Buffer D: 10% (v/v) グリセロール,50 mM HEPES pH 7.5,100 mM \(\ce{NaCl}\),1 mM \(\ce{CaCl2}\),0.003% (w/v) LMNG
- Buffer E: 30% (v/v) グリセロール,50 mM HEPES pH 7.5,100 mM \(\ce{NaCl}\),1 mM \(\ce{CaCl2}\),0.003% (w/v) LMNG
下記の操作で作成するグリセールベッドはサンプル用と遠心時のバランス用に2本用意する。超遠心用のチューブ(13 ml 容量)に 5.25 ml の水を加え、チューブ外側に水位をなぞるようにマジックペンで印をつける(図3)。チューブから水をよく取り除き、Buffer D を 5.25 ml の水位になるまで加える。さらに、Buffer E を針の長い注射器を用いてチューブ底からゆっくりと加える。チューブにパラフィルムでよく蓋をして静かに横に倒し、4℃で2時間静置する。チューブを静かに縦に直してパラフィルムを取り外す。
2本用意したグリセールベッドの一方に、オリゴマー化処理をしたサンプル約 1 ml をピペットマンによって静かに積層する。もう一方のチューブには重さ合わせのために水を積層するなどした後、厳密にチューブ二本の重さを合わせる。P40ST(スイングローター)を用いて、4℃、230,139 ×g で16時間超遠心する(図4)。
2. フラクション分け
サンプルを加え超遠心を終えたチューブの底に、シリンジの針を用いて孔を開ける(図4)。開けた孔から滴る水滴を5滴ずつ1フラクションとして、チューブ内の溶液が無くなるまで回収する(およそ1フラクションあたり 250 μl となる)。回収したフラクションをそれぞれナノドロップによって濃度(A280nm)を計測する。フラクション番号を横軸に濃度(A280nm)としてプロットしてグラフを作成する。各ピークを SDS-PAGE して Ib 膜孔が含まれるフラクションを同定し、1つの新しいチューブにまとめる。
3. Buffer 交換
Buffer F: 10 mM HEPES pH 7.5,1 mM \(\ce{CaCl2}\),0.003% (w/v) LMNG を用意する。密度勾配遠心由来のグリセロールを除去するために、PD10 を3回通して Buffer F に交換する。さらに Amicon 100K を用いて 2 mg/ml 程度に濃縮する。サンプルを液体窒素によって瞬間凍結し、−80℃で冷凍保存する。
Ia の調製
イオタ毒素(Ia・Ib)を含む二成分毒の酵素成分は N 末端側から透過していくと考えられたため、N 末端になるべく不要な残基が付与されることを避けたいと考えた。このため Ia の精製には C 末端に付与した His タグを使用した。
1. クローニングおよび培養
Ia 遺伝子(Uniplot ID:Q46220)が導入された pET-21b ベクターを用意し、これを大腸菌 E.coli BL21 (DE3) にヒートショック法で形質転換させる。これをアンピシリン(50 μg/ml)を含む LB 固体培地に塗布し、37℃で一晩培養する。翌日、固体培地上に形成されたコロニーを用いて、アンピシリン(50 μg/ml)を含む 3 ml の LB 液体培地に移し、37℃の温浴槽で一晩震盪培養する。培養された菌体液とあらかじめオートクレーブされた50% (v/v) グリセロール溶液と混合し、これをグリセロールストックとして−80℃で冷凍保存する。
少量のグリセロールストックをアンピシリン(50 μg/ml)を含む 3 ml の LB 液体培地に加え、37℃の温浴槽で一晩震盪培養する。翌日、菌体液全量をアンピシリン(50 μg/ml)を含む 1 L の LB 液体培地に加え、37℃で震盪培養する。LB 液体培地をベースラインとして、培養液の濁度(OD600)=1.5(AU)となったところで培養を一度止め、培養液フラスコを氷水で冷やす。十分に冷却された培養液に終濃度 0.5 mM となるように IPTG を加え、37℃で一晩震盪培養する。培養液を4℃、12,092 ×g で5分間遠心分離する。沈降した菌体を 50 ml チューブにスパーテルなどで移し替え、-80℃で冷凍保存する。
2. Ia の精製
あらかじめ下記の Buffer を用意する。
- Buffer G: 20 mM Tris pH 8.0,300 mM \(\ce{NaCl}\),20 mM イミダゾール
- Buffer H: 500 mM Tris pH 8.0,300 mM \(\ce{NaCl}\),500 mM イミダゾール
- Buffer I: 20 mM Tris pH 8.0,2 mM \(\ce{CaCl2}\)
- Buffer J: 20 mM Tris pH 8.0,1 M \(\ce{NaCl}\),2 mM \(\ce{CaCl2}\)
- Buffer K: 10 mM Tris pH 8.0,100 mM \(\ce{NaCl}\)
冷凍保存した菌体を室温で融解し一錠の cOmplete を溶解させた 120 ml の Buffer G に懸濁する。これを金属製のカップに移し、氷水上でソニケーションによって大腸菌を破砕する。破砕菌体液を超遠心用のチューブに移し替え、4℃、180,000 ×g で40分間、超遠心にかける。上清をあらかじめ Buffer G に置換された体積 5 ml の Ni-NTA 樹脂と4℃で1時間転倒混和する。樹脂からフロースルー回収後、Buffer G を用いて1フラクション 5 ml の wash を行う。ナノドロップによって wash 画分の濃度(A280nm)を計測し、これが 0.1 mg/ml を下回るまで wash を繰り返す。Buffer H を用いて1フラクション 1 ml の Elution を行う。溶出されたタンパク質の濃度(A280nm)がピークを超えた後に 0.1 mg/ml を下回るまで Elution を繰り返す。得られた Elution 画分を集め、Amicon 30K によって Buffer I ヘの交換を行う。さらに陰イオン交換樹脂(HiTrap Q HP 5ml column)を用いて、Buffer I と J よる \(\ce{NaCl}\) 濃度勾配によって Ia を溶出する。得られた Ia を含む溶液を Amicon 30K によって、Buffer K への置換と濃縮を行う。濃縮されたサンプルは 500 μl 程度になるように分注し、液体窒素による瞬間凍結の後、−80℃で冷凍保存する。
複合体の形成およびグリッド作製
密度勾配遠心前に加え複合体にしてしまうと、一連の精製過程で Ia が Ib 膜孔から解離してしまいサンプル中の複合体の割合が著しく減少してしまった。この結果を受けて、最終的に、密度勾配遠心で精製した Ib 膜孔に対して十分量のIaを加えて電子顕微鏡による観察を行った。
1. 複合体形成
密度勾配遠心法によって得られた Ib 膜孔にモル比で3倍量の Ia を加える。
2. グリッド作製
Buffer L: 10 mM HEPES pH 7.5,1 mM \(\ce{CaCl2}\) を用意する。Buffer L で希釈したサンプル 2.6 μl を、グロー放電によって親水化処理した Quantifoil R1.2/1.3 に100%湿度下でアプライする。濾紙で4.5秒間の吸水を行い液体エタンによって瞬時に凍結する。凍結したグリッドは適宜、各々のクライオ電子顕微鏡で観察・データ測定を行う。
工夫とコツ
Ib の膜孔形成
1. クローニングおよび培養
菌体を培養する培地は必ず抗生物質の添加前にオートクレーブ滅菌する。同様にグリセロールストック作成に使用する50% (v/v) グリセロール溶液もオートクレーブする。凍結したグリセロールストックはなるべく融解しないように、使用したらすぐに冷凍庫に戻す。数ヶ月に一度新たにグリセロールストックを作成すること。
2. Ib 膜孔の精製
DTT などの還元剤は Buffer の使用直前に加える。タンパク質を扱う全ての工程はできる限り氷上で行うなど、温度が上がらないようにする。サンプルの保存は凍結がなるべく迅速になるように 500 μl 以下で保存すること。
3. オリゴマー形成
Ib は Chymotrypsin 処理のみではオリゴマー形成効率が極めて低いが、エタノールの添加によってオリゴマー形成効率が飛躍的に向上する。基本的に10% (v/v) のエタノールがあればほぼ全ての Ib がオリゴマーになるが、精製ロットによって若干オリゴマー化効率に差異がある(原因は不明)ので、新たに精製した Ib は0–20% (v/v) のエタノール条件でオリゴマー化効率をスクリーニングすること。Ib 膜孔は SDS 耐性があるため、SDS-PAGE をしても解離せずオリゴマーとしてバンドを確認することができる(図2.b)。
Ib 膜孔の精製
1. 密度勾配遠心
超遠心に用いるチューブは必ず対応可能な遠心力を確認すること(物品リストに書いたチューブであればこのプロトコルにおいて問題ない)。一方のグリセロール溶液にCBBを加えるなどして着色してみると、起こした時に勾配が作られていることを可視化することができる。グラジエントメーカーがある研究室ではそちらを使っても問題ない。作成したグリセロールベッドは勾配が崩れないように、超遠心機まで慎重に運ぶ。スイングローターはサンプルを入れたチューブとそのバランスコントロールの重さが釣り合うようにセットする。チューブを入れていないバケットも全てローターに取り付けて遠心を行う。グリセロール濃度勾配や遠心力によって分離の様子が変わるため、各自のサンプルの分子量によって条件を最適化することが好ましい。
当初の最適な LMNG 濃度がまだわからなかった頃の実験から、試料中の過剰な LMNG の存在は電子顕微鏡画像上に LMNG のミセルと思われる像を出現させてしまうことがわかった(図5)。この像は希釈をすることで減少するが、同時に画像あたりの粒子数も減らしてしまう。最終的に Ib 膜孔では0.003% (w/v) で最適な画像を撮影できることがわかったが、扱うタンパク質によって LMNG 濃度の最適化は必ず行うこと。
2. フラクション分け
フラクション分けはチューブ底に孔を開け、漏れ出た水滴を集めるアナログな方法で行う。市販の注射針の先端をペンチで根元から数 mm になるように捻じ切って尖らせると孔を開けやすい。ペンチで捻じ切って取れた針の先は怪我しないように気をつけて、適切に処理する。人間の手で行うことから孔の大きさが毎回微妙に異なるため、目的タンパク質のフラクション位置に微妙なズレが生じる。ただの水をチューブに入れて孔を開ける練習をあらかじめしておくとよい。またチューブ内には超遠心によって分離した溶液が入っているため、一連の作業でこれを崩さぬように気を付ける。
3. Buffer 交換
特記事項なし。
Ia の調製
1. クローニングおよび培養
Ib のクローニングおよび培養に同じ。
2. Ia の精製
Ia は凍結融解をすると白く濁ることがあった。可能であれば精製したばかりの凍結保存をしていない Ia を使用すること。
複合体の形成およびグリッド作製
1. 複合体形成
精製した Ib 膜孔に mol 比3倍量の Ia を加えることで、非結合の Ia も試料中には存在するが、Ia は 50 kDa 以下の小さなタンパク質であるため、解析にはあまり悪影響しないと考えられる。単粒子解析にて二次元平均画像を作成すると、Ib 膜孔の上部に Ia が結合したと見られるボヤけた密度が確認できる(図6)。
2. グリッド作製
サンプルの希釈系列は数段階用意しておき、電子顕微鏡で観察しながら最適な濃度を決めるのが好ましい。経験的に0.003% LMNG (w/v) 存在下で 2 mg/ml 程度のサンプル濃度であれば、十分に単分散し高頻度で粒子を観察できるグリッドを作製可能である。
実験の安全
本実験では形質転換体の大腸菌を扱うため、培養に使用した容器や器具はオートクレーブなどで十分に滅菌処理を行う。研究室外への形質転換体の漏出が無いように最大限の注意を払って実験を行う。シリンジの針なども扱うのでケガのないように使用・廃棄について注意する。短い針の作成には必ずペンチを使用すること。ハサミで切ろうとすると先が飛んでしまい非常に危険である。グリッド凍結に用いる液体エタンの付着による火傷には安全メガネをかけるなど十分に配慮すること。
終わりに
本プロトコルの作成にあたって行ってきた実験にたくさんの方々の惜しみ無いご協力をいただきましたこと、この場を借りて感謝申し上げさせていただきたいと思います。試料調製についてたくさんのディスカッションを交わしていただき、吉田徹先生、津下英明先生、誠にありがとうございました。光岡薫先生、川本晃大先生、岩崎憲治先生には電子顕微鏡による観察・解析におけるご援助をいただきましたこと、心より感謝申し上げます。
文献
- Yamada, T. et al., Nat. Struct. Mol. Biol. 27, 288–296 (2020)
- Barth, H. et al., Microbiol. Mol. Biol. Rev. MMBR 68, 373–402 (2004)
- Lacy, D. B. et al., Proc. Natl. Acad. Sci. U. S. A. 101, 13147–13151 (2004)
- Gibert, M. et al., Infect. Immun. 68, 3848–3853 (2000)
- Miller, C. et al., Biochemistry 38, 10432–10441 (1999)
- Jiang, J. et al., Nature 521, 545–549 (2015)
- Simpson, L. et al., Infect. Immun. 55, 118–122 (1987)
- Tsuge, H. et al., J. Mol. Biol. 325, 471–483 (2003)
- Mio, K. et al., Biophys. Rev. 10, 307–316 (2017)
- Rouck, J. E. et al., FEBS Lett. 591, 2057–2088 (2017)
- Hauer, F. et al., Structure, 23(9), 1769–1775 (2015)
-
図1:Ia–Ib 膜孔複合体の構造
Ia–Ib 膜孔複合体のモデル(PDB ID:6klw)。Ib 膜孔は Ib-pore として表記している。 -
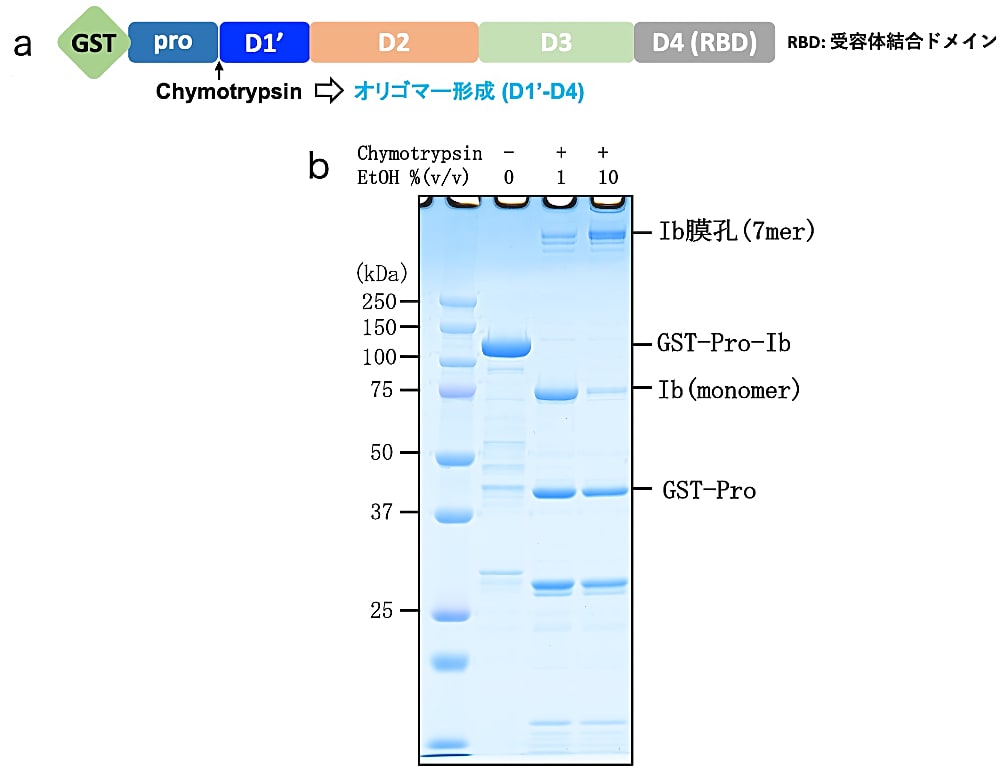
図2:Ib 膜孔のオリゴマー形成
(a)Ib のコンストラクトマップ。chymotrypsin 処理によって Pro 配列とともに GST タグが切断される。(b)Ib 膜孔のオリゴマー形成を非加熱 SDS-PAGE で確認した結果。Chymotrypsin 処理後、エタノール(EtOH と表記)を加えることでモノマーからオリゴマーへの形成が促進されることがわかる。 -
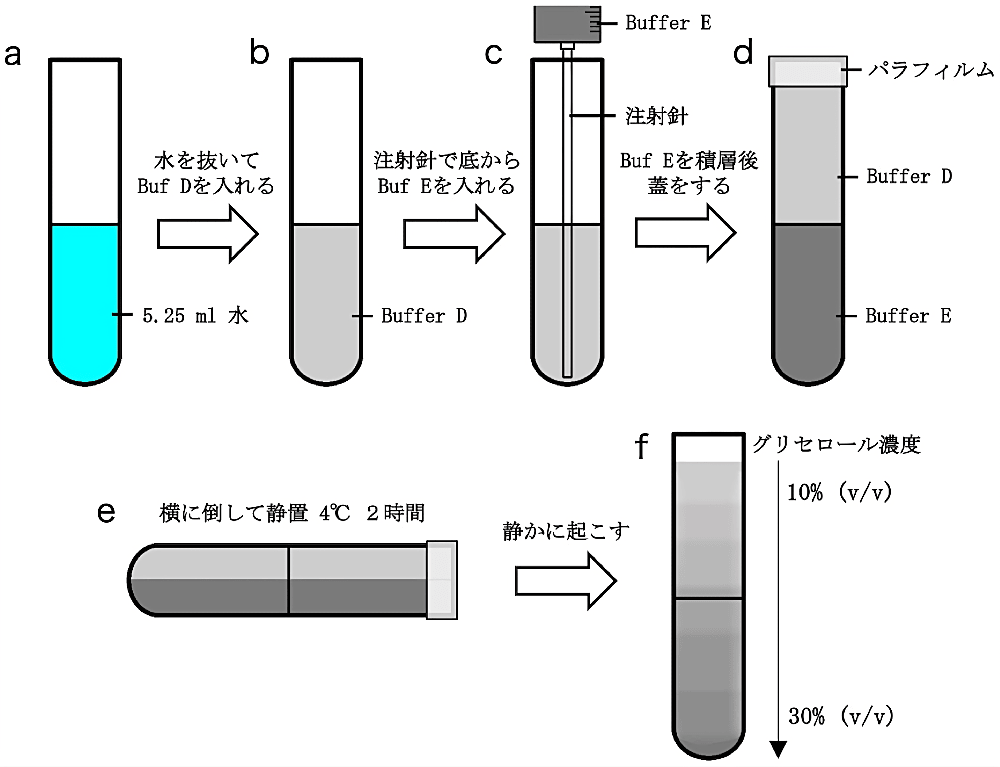
図3:密度勾配遠心の準備
(a)水を 5.25 ml 入れて水位をペンでなぞる。(b)ペンでつけた目印まで Buffer D を入れる。(c)ペンでつけた目印まで Buffer E を入れる。(d)パラフィルムで蓋をする。(e)横に倒して静置する。(f)再び起こすとグリセロール濃度勾配が形成されている。 -
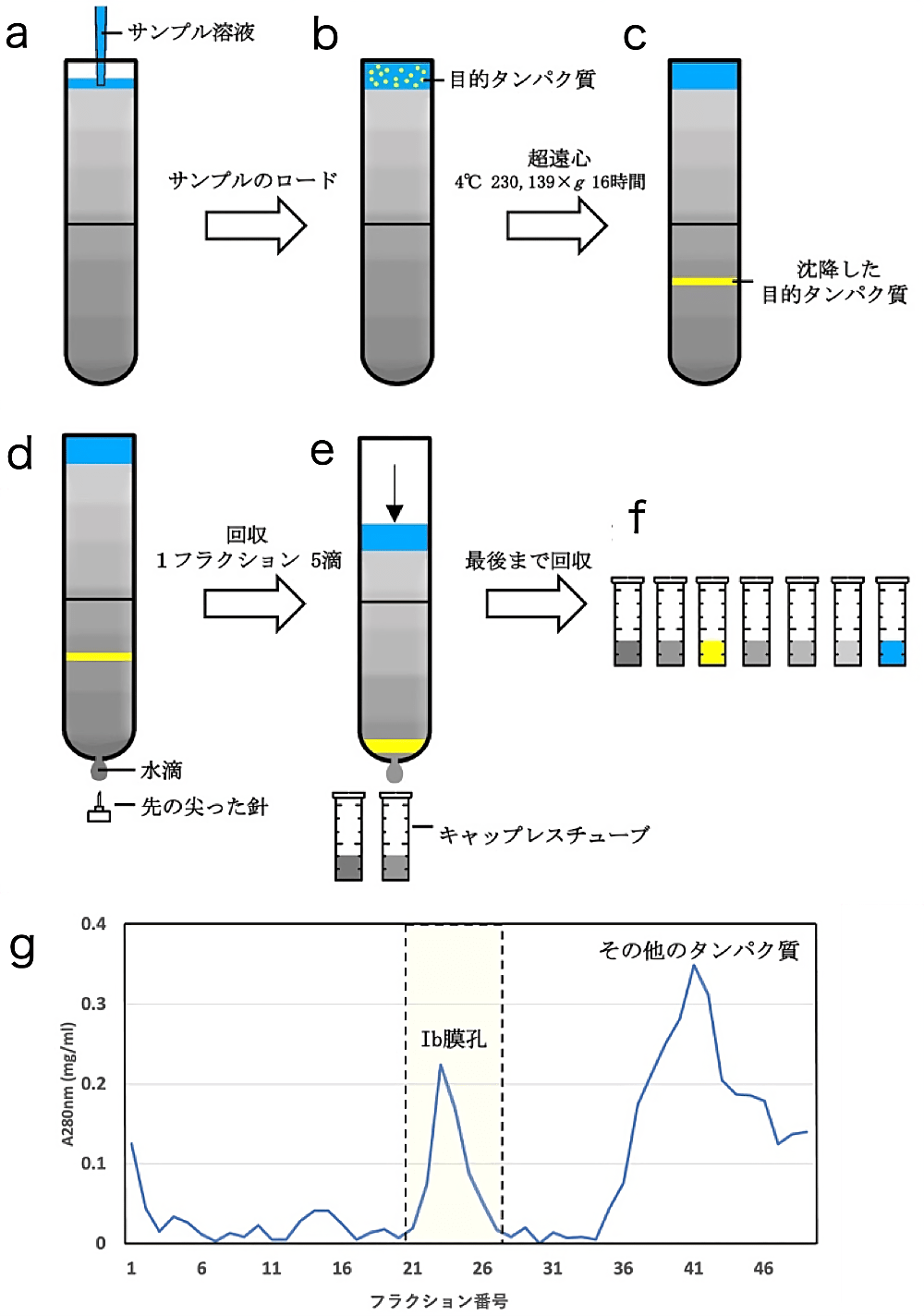
図4:密度勾配遠心
(a)グリセロールベッドにサンプル溶液を積層する。(b)超遠心にかける(c)超遠心後、分子量に応じた位置にタンパク質が沈降している。(d)チューブ底に針で孔を開ける。(e)漏れ出す水滴を集めフラクションとして回収する。(f)チューブが空になるまで回収する。(g)回収したフラクションのタンパク質濃度の計測を行い作成したグラフ。Ib 膜孔サンプルの場合、最初のフラクションに凝集した蛋白質、中位に Ib 膜孔、終盤であまり沈降しなかった小さなタンパク質のピークを確認できる。 -

図5:LMNG由来の像による画像への影響
Talos Arctica(200 kV)で撮影した電子顕微鏡画像。a,b の試料は0.01% (w/v) LMNG を含む同じ Ib 膜孔サンプル(1.9 mg/ml)を、LMNG を含まない buffer で異なる希釈度に薄めたもの。(a)1/2希釈した試料。紐状の LMNG ミセルと思われる像が大量に見える。(b)1/5希釈した試料。紐状の像があまり見えなくなる。 -
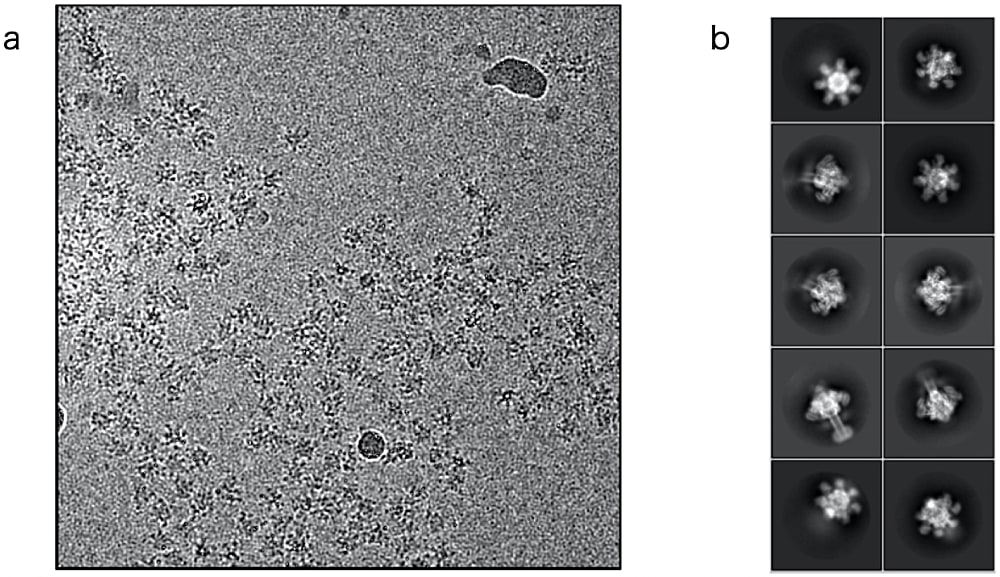
図6:Ia–Ib 膜孔複合体の電子顕微鏡画像
(a)Titan Krios(300 kV)で撮影した電子顕微鏡画像。よく単分散した様子を確認できる。(b)電子顕微鏡画像から抽出した粒子の二次元平均像。
