

概要
分子動力学法(Molecular Dynamics, MD)は、原子・分子間に働く力を用いて運動方程式を数値的に解くことで、粒子の運動を求める方法である。揺らぎや運動などのダイナミクス解析、自由エネルギー面の探索などに利用されている。近年は、計算機と計算手法の発展により、計算化学の専門家だけでなく実験研究者にもモデリング、リファインメント、創薬・分子デザインに広く利用されるようになっている。本稿では、理化学研究所計算科学研究センター粒子系生物物理研究チームを中心に開発されている MD ソフトウェア GENESIS(フリーソフトとして公開中)を用いて、水溶性タンパク質の MD 計算を行う方法を述べる。具体的例を挙げ、GENESIS のインストール、計算するタンパク質分子モデルの準備、MD 計算(予備計算と本計算)を概説する。
イントロダクション
分子動力学法(Molecular Dynamics, MD)は、原子・分子間に働く力(相互作用力)を用いて運動方程式を解くことで、粒子の運動を求める方法である。相互作用する多粒子の運動方程式は数値解法と必要とする。相互作用力の計算には、タンパク質、核酸などの生体高分子を対象とする「力場(force field)」と呼ばれる経験的な関数を用いる。力場による相互作用力計算と運動方程式による時間発展計算には、コンピュータの高い演算能力が大いに威力を発揮する。近年の計算機の発展(特にグラフィックカード(Graphics Processing Unit, GPU)による高速演算)と様々な手法の開発により、計算可能なシステムサイズ、時間オーダーが飛躍的に上がっている。開発された手法の多くは様々な MD ソフトウェアに導入され、利用可能となっている。本稿では、その一つである理化学研究所、計算科学研究センター、粒子系生物物理研究チームを中心に開発されている GENESIS [1,2] を用いて水溶性タンパク質のMD 計算を行う方法を概説する。
装置・ソフトウェア
GENESIS によるシミュレーションの実行には、ソフトウェア、計算機(ワークステーション)、ソフトウェアを動作させるための開発環境が必要となる。近年、多くの大学、研究所の計算機センターではトライアルユースなどが安価(もしくは無料)で提供されている。GENESIS に必要な開発環境は、筆者が知る限り多くの計算機センターで標準的に導入されているものである。また、自然科学研究機構岡崎共通研究施設計算科学研究センターのスーパーコンピュータでは GENESIS が導入されている。計算機のメンテナンスコストを考えると、計算機センターの活用も推奨したい。更に結果を確認するための可視化ソフトウェアが必要となる。
- GENESIS(フリーソフトとして公開中)
最新版は GENESIS のウェブサイトから取得可能。 - 本稿で紹介する入力ファイル一式は、ここから入手可能である。ファイルを以下のように展開する。
$ tar xvhf Protocol_ howtoGENESIS.tar.bz2 $ cd Protocol_ howtoGENESIS/Protocol_ howtoGENESIS/には以下のディレクトリが存在する。1_minimization/ 2_A_heating/ 2_B_NVT/ 2_C_NPT/ 3_equilibration/ 4_production/ molecule_files/ toppar/ - 可視化ソフトウェア: 分子の構造、運動(トラジェクトリ)を確認する。Illinois 大学 Urbana-Champaign 校で開発されている VMD [3] の利用を推奨する。このソフトウェアは、GENESIS や他の MD ソフトウェアで利用されるファイル形式(dcd)を直接読む事が可能。VMD は Linux/Windows/MacOS X にも対応
自分で計算機環境を整える場合は以下が必要となる。
- ワークステーション
- CPU: Intel, SPARC64(HPC-ACE), AMD(Opteron)が動作確認済
速度上から Intel Xeon SandyBridge 世代以降を推奨 - メモリ: 20万原子(注)の計算を1ノードで行うにはソフトウェアに 10 GB 程度の割り当てが必要となるため、1ノード 32 GB 以上が望ましい
- CUDA 対応の GPU カード(NVIDIA)(任意): Compute Capability 3.5, 3.7, 6.0, 6.1, 7.0 が動作確認済
- CPU: Intel, SPARC64(HPC-ACE), AMD(Opteron)が動作確認済
- オペレーションシステム(OS)、開発環境(コンパイラ、ライブラリ)
- Unix 系 OS: Linux, Free BSD, MacOS X などが利用可能
- C、Fortran コンパイラ: gcc, gfortran ならversion 4.4.7 以降、Intel Fortran/C なら version 12 以降
- Message Passing Interface(MPI): OpenMPI(1.10 以降推奨), Intel MPI, 富士通 MPI が動作確認済。OpenMPI は Unix 系OS の多くではパッケージ化されているため、システムツール(apt, dnf/yum, dpkg など)からインストール可能
- Basic Linear Algebra Subprograms(BLAS)、Linear Algebra PACKage(LAPACK)ライブラリ: Unix 系 OS の多くではパッケージ化されているため、システムツールからインストール可能。また、Intel MKL, 富士通 SSL の利用可能
- GNU autoconf: version 1.3 以降
- (GPU 利用の場合のみ)CUDA ライブラリ、コンパイラ: version 8.0 以降
注:分子モデルには溶媒分子も含まれる。20万原子は大体、分子量 60 – 80 k のタンパク質の分子モデルに対応する。ただし、後述のようにタンパク質の形状や性質によりモデルのサイズ(溶媒分子の数)は大きく変化することに注意されたい。
実験手順
- タンパク質分子モデルの準備(セットアップ)
- 構造最適化
- 温度と圧力(体積)の調整
- 平衡化
- 本計算
実験の詳細
GENESIS の内容物
GENESIS は、2つの MD エンジン(spdyn, atdyn)と複数の解析用ソフトウェアからなる。spdyn(SPartial decomposition DYNamics simulation)は超並列計算のために設計され、単精度計算、グラフィックカードの利用が可能である。一方、atdyn(ATomic decomposition DYNamics simulation)は利用者による機能追加が可能なように平易な内部構造となっている。CHARMM, AMBER などの全原子モデルを利用する場合は spdyn を推奨する。本稿は spdyn の利用について記載する。
GENESIS のインストール
入手した GENESIS の tar.bz2 ファイルを展開し、以下の要領でインストールする。実行ファイルは genesis ディレクトリ直下に bin/ ディレクトリに作成されるため、特権ユーザ(super user, root)になる必要はない。
$ tar xvhf genesis-1.3.x.tar.bz2
$ cd ./genesis-1.3.x
$ ./configure
$ make ‒j 4 install
bin/ ディレクトリに spdyn, atdyn と解析用のソフトウェアがインストールされる。
GPU を利用する場合は、以下の手順でインストールを行う。
$ tar xvhf genesis-1.3.x.tar.bz2
$ cd ./genesis-1.3.x
$ ./configure --enable-single --enable-gpu
$ make ‒j 4 install
configure はコンパイル時のオプションを与える機能がある。GENESIS は多くの場合自動的に設定を行うが、手動で設定を行う事もできる。configure で利用可能なオプションは以下のコマンドで確認できる。
$ ./configure --help
正しくインストールされたかを確認するために、テストを行う。テストセット(tests-1.3.x.tar.bz2)も GENESIS のウェブサイトから入手可能である。
$ tar xvhf tests-1.3.x.tar.bz2
$ cd tests/regression_test
sh/bash の場合は
$ export OMP_NUM_THREADS=1
csh/tcsh の場合は
$ setenv OMP_NUM_THREADS 1
test.py を用いて以下のように実行する。
./test.py “mpirun -np 8 実行ファイルの絶対パス/実行ファイル”
例えば、/home/user/genesis にインストールされている場合は、以下となる。
$ ./test.py “mpirun -np 8 /home/user/genesis/bin/spdyn”
また、GPU を利用する場合は gpu オプションを付ける
$ ./test.py “mpirun -np 8 /home/user/genesis/bin/spdyn” gpu
上を実行すると、用意された複数のテストセットが順番に実行される。各テストが正常に動作すると以下のように ”Passed” が表示される。
(例)
Checking ./test_common/dna/CUTOFF
Checking diff between ref and test...
Passed (tolerance = 3.00e-05(ene), 3.00e-03(virial))
また、全てのテストセットの最後に以下のように何個のテストが成功したかが記載される。 テストの総数はテストの種類、GENESIS のバージョンや GPU の有無によって変化する。
(例)
Passed 44 / 44
Failed 0 / 44
Aborted 0 / 44
重要な点としては、このテストは MPI 並列数が8で実行することを想定して作成されているため、必ず MPI 並列数は8で行わなければならない。
CHARMM-GUI を使ったタンパク質分子モデルの準備
次にタンパク質分子モデルの準備(セットアップ)を行う。セットアップを行うツールはいくつか公開されており、本稿では Web ブラウザベースでセットアップができる CHARMM-GUI [4] を利用する方法を紹介する。このソフトウェアは Lehigh 大学の Im 教授のグループで開発されており、セットアップを行い MD の実行ファイルに必要な入力ファイルを出力する。以下に、MD のセットアップを行い、GENESIS に必要なファイルを作成する方法を示す。
- CHARMM-GUI のウェブサイトにアクセスする
- 左側のメニューから Input Generator を選ぶ。
- 左側のメニューから Quick MD Simulator を選ぶ。
- 下側の入力ボックス内で、PDB の ID もしくは、PDB のアップロードを選ぶ。本稿ではテストとして RCSB の PDB からユビキチンの PDB(1UBQ)を指定する(図1)。
- PDB の情報を確認する。本稿の例では入れていないが、Water のチェックボックスにチェックを入れると結晶水もシステムに含まれる(図2)。
- 変異の導入、プロトン化状態(Asp, Glu, Lys, His)の変更・設定、リン酸化や蛍光色素の導入など、タンパク質の情報を変更したい場合には、チェックボックスに適宜チェックを入れる。今回は特に設定を行わない。
- 溶媒水分子、イオンの追加。水分子を詰めたボックスにタンパク質とイオンを入れる方法をとる。このボックスのサイズがシステムのサイズとなる。体積に比例して計算コストは増大する(粒子数の1~2乗)。計算時間を抑えるためには、デフォルトであるタンパク質の端からシミュレーションボックスの端まで10 Åの余裕を持たせる設定が妥当である。ただし、周期境界条件で計算を行うため、この値が小さいとボックス越しに自分同士と相互作用してしまうため、十分に注意が必要となる。特に、Fold/Unfold 間の転移などタンパク質の構造が大きく変化する場合は、タンパク質が取りうる最大のサイズを考えてシステムのサイズを決めなければならない。また、GENESIS ではボックスの形は rectangular のみが可能である。導入するイオンの数は、対象によって適切に設定する(本稿の例ではデフォルトである0.15 M KCl を使用)。ただし、長距離相互作用計算で広く用いられている Particle Mesh Ewald(PME)法での計算では、電荷(net charge)は0でなければならない事に留意されたい。そのため、イオン濃度を設定しない場合でも Add neutralizing ions を選定する(図3)。この過程は5–10分ほど掛かる。
- システムの確認画面となる。システムのサイズ、イオンの数などを確認する。下の Periodic Boundary Condition Options に関しては、特別の理由がない限り変更させない(図4)。
- 出力する形式の選択画面となる。GENESIS を選択。
- セットアップは完了となる。右端の “Download” ボタンを押し、charmm-gui.tgz というファイルを保存する。
- 以下の要領で charmm-gui.tgz を展開し、ディレクトリ内へ移動する。
$ tar xvhf charm-gui.tgz $ cd charmm-gui - 展開された
charmm-guiディレクトリには大量のファイルが存在するが本稿では以下のファイルのみを抜き出して利用する。
①力場ファイル: 力場計算に必要な情報を与える。CHARMM 力場では、それぞれの分子の定義を記載した topology(top)ファイル、原子の質量、電荷、原子間の相互作用のパラメータを記載した parameter(par)ファイル、追加パラメータ(top と par の両方)の情報を記載したstream(str)ファイルからなる。CHARMM の力場の場合は、タンパク質、核酸、糖質、脂質膜分子等の分子種毎にファイルが分かれている。toppar/には現在公開されている全分子種用のファイルがある。全てを読みこませても問題はないが、本稿で説明する水溶性タンパク質の計算ではtop_all36_prot.rtf,par_all36m_prot.prm,toppar_water_ions.strのみが必要である。
②protein structure file(psf)ファイル: タンパク質分子モデルの情報(原子数、残基や原子の種類、相互作用の情報)が記載されている。
step3_pbcsetup.xplor.ext.psf
③pdb ファイル: タンパク質分子モデルの構造(座標)情報が記載されている。
step3_pbcsetup.pdb
GENESIS による MD 計算
A. GENESIS のコントロールファイル
GENESIS では、コントロールファイルと呼ばれるファイルを読み、MD 計算の設定を行う。コントロールファイルでは、主に以下のようなセクションに分けられ、各パラメータが設定されている。
- [INPUT] 入力ファイルの情報(力場ファイルなど)
- [OUTPUT] 出力ファイルの情報(トラジェクトリファイルなど)
- [ENERGY] 力場計算の情報(力場の種類や静電相互作用の計算手法など)
- [DYNAMICS] 時間幅や数値積分の方法の情報
- [MINIMIZE] 構造最適化計算の情報(構造最適化計算のみ利用)
- [CONSTRAINT] 分子内拘束(SHAKE/RATTLE, SETTLE)の情報
- [ENSEMBLE] アンサンブル計算の情報(温度や圧力など)
- [BOUNDARY] シミュレーションボックスの情報(サイズや周期境界条件など)
- [RESTRAINTS] 拘束(restraint)ポテンシャルの情報
- [SELECTION] 拘束計算などに利用する原子・分子グループの情報
また、spdyn/atdyn などの MD プログラム、解析プログラムを以下のように実行することでコントロールファイルのテンプレートが作成される。
$ 実行ファイルのパス/実行ファイル ‒h ctrl > コントロールファイルのテンプレート
出力されたテンプレートは、vim や emacs などのエディターを使い修正し、コントロールファイルを作成する。
B. GENESIS の実行方法
GENESIS は以下のように実行される。
mpirun -np 8 実行ファイルのパス/spdyn コントロールファイル > ログファイル
ログファイル内の出力は7段階(STEP 0-6)に分かれており、それぞれ以下の情報が出力される。
- STEP 0: 計算環境(ハードウェア、ソフトウェア)
- STEP 1: コントロールファイルでの入力パラメータ
- STEP 2: 並列数(プロセス、スレッド数)
- STEP 3: 分子・エネルギー関数情報
- STEP 4: 初期座標でのエネルギー値
- STEP 5: 各ステップでの各エネルギー値、温度、体積などのデータ
- STEP 6: 演算時間
STEP 5の各ステップのデータの出力と並んで、STEP 3の分子・エネルギー関数情報の出力は重要である。この出力と、コントロールファイルにて設定したパラメータや分子、力場ファイルが整合しているかを確認されたい。ここに設定した物と異なるパラメータ、ファイルが出力されている場合は、意図しない計算を行う場合がある。その場合は、出力内の警告(WARNING)を参考に適宜修正を行う。
C. GENESIS によるMD 計算の流れ
MD の計算は、一般的には以下の手順で実行される。
- 構造最適化
- 温度と圧力(体積)の調整
- 平衡化
- 本計算
手順1–3がタンパク質分子モデルを温度、圧力などの計算条件に合わせる予備計算である。人為的な手法で作られた初期構造は、原子間の距離が近づき過ぎ、不自然な原子配置を持つことが少なくない。不自然な原子配置では、特定の原子間の相互作用力が大きくなり、数値解の誤差が増大し、粒子の運動を正しく求められない。その場合は、分子の一部のみが大きく動き、最悪の場合はタンパク質分子モデル全体が崩壊することもある。このような「不安定なシミュレーション」を避けるため、予備計算を慎重かつ十分に行う事が必要である。
CHARMM-GUI から出力されるコントロールファイルはミニマルな仕様となっているため、本稿では著者が作成したファイルを例に説明を行う。
1. 構造最適化(ディレクトリ 1_minimization/)
構造最適化は予備計算の第一段階として、相互作用エネルギー値が下がる方向に粒子を動かし、不安定な原子配置を取り除く計算である。GENESIS では現在、最急降下法(Steepest decent method)が利用可能である。図5に GENESIS のコントロールファイルを示す。
この段階では不安定な原子配置が存在するため、計算のターゲットになるタンパク質・高分子・リガンドには位置や距離に拘束を適宜導入して、極端に分子が動かないようにすることが必要である。ステップ数は、あくまで極端にエネルギーが高い構造をある程度解消することが目的であるため、数千ステップ程度で良い。GENESIS では、不自然な距離にある原子対を出力し、かつ、相互作用力の値に制限をかける “contact_check” というオプションが搭載されている。構造最適化や温度・体積を調整するシミュレーションでは利用されたい。数千ステップ後でも、エネルギー値が上下するなど落ち着かない場合は、「工夫とコツ」(後述)の「タンパク質分子モデルの確認時の注意」を参照し、構造の確認を行う。
2. 温度と圧力(体積)の調整
システムを計算する標的温度、圧力に慣らす。安定した本計算を行うためには最も重要なステップである。温度上昇、温度一定、体積調整の3段階の計算を行う。
A) 温度上昇シミュレーション(ディレクトリ 2_A_heating/)
温度上昇シミュレーションは、構造最適化によりある程度安定化された構造に対して、システムの温度をゆっくりと上昇させ、標的温度に至らせるものである。GENESIS では、“Annealing” オプションを使うことが可能である(図6)。少しずつ標的温度を上昇させながら複数回シミュレーションを行っても良い。
B) 温度一定シミュレーション(ディレクトリ 2_B_NVT/)
前項での温度上昇シミュレーションでは、標的温度での計算は500 step しかできず、システムの温度の調整としては不十分である。そのため、体積調整シミュレーションを行う前に温度一定(NVT)のシミュレーションを行う。この段階では、ログファイルから温度の時間変化を確認し、設定した温度の周辺を揺らいでいることを確認する。体積が変化しないため、シミュレーションボックス内に真空のバブルができることがある。これは、人為的に作成したシミュレーションボックスは、分子の密度が必ずしも正しくないためである。このバブルは次のステップで解消させる。
C) 体積(密度)調整シミュレーション(ディレクトリ 2_C_NPT/)
人為的に作成したシミュレーションボックスをシステムに最適な密度に合わせるためにNPT 計算をおこなう。特に本計算がNVT 条件の場合では、温度上昇、温度一定シミュレーションの後に直接平衡化を行うと、シミュレーションボックス内のバブルが解消されない。そのため、平衡化の前に必ず1~2 ns 程度の NPT 計算を行い、最適な密度に合わせる必要がある。体積の時間変化を確認し、体積がある値の周辺で揺らぐ状態にある(ドリフトしない)事を確認する。また、圧力はステップ数毎の揺らぎが大き過ぎるため、この場合の指標としては適切ではない。体積変化により位置拘束の力が急速に上がり、粒子の運動や圧力の計算が正しく行われなくなる恐れがあるため、基本的には位置拘束は使わない。
3. 平衡化(ディレクトリ 3_equilibration/)
本計算で利用する条件と同じ条件でシステムを安定化すべく、同じ条件での計算を行う。これ以降の計算では、contact_check オプションは必ず外すこと。2 fs 以上の時間幅や長距離相互作用計算や温度・圧力制御を数ステップ置きに行う Multiple time step 法などの高速化スキームは、この段階から使用する。最低でも 20 ns 程度の計算が必要となる。これは、20 ns で十分という意味ではない。平衡化に要するステップ 数は計算するシステムによって大きく異なるため、温度、体積(NPT 条件の場合)、ポテンシャルエネルギーの時間変化がドリフトしていない事を確認する。
4. 本計算(ディレクトリ 4_production/)
実際にデータを取るための計算である。サブ μs から、最近では数十 μs オーダーの計算も珍しくなくなった。粒子の軌跡をトラジェクトリとして書き出しを行うことで解析を行い、様々な原子・分子の性質を計算することが可能となる。チュートリアルのコントロールファイルでは例のために 20 ns となっているが、実際の研究ではもっと長時間の計算が必要であることを強調したい。
MD 計算は、極小値に向けて値が収束していく計算と異なり、出力される温度、体積、エネルギー値などの数値データだけではその成否を判断できない。そのため、本計算だけでなく予備計算の段階から、分子たちが意図した条件内で運動している事を、タンパク質の揺らぎ(RMSF)、初期座標からの RMSD、リガンド、残基間の塩橋や水素結合ネットワークの構造情報を随時計測し、その時間変化を丁寧に追うことも必要となる(図7)。
D. 可視化ソフト(VMD)による MD 計算のトラジェクトリ表示
VMD を用いてトラジェクトリを可視化するためには、分子情報(原子や残基の名前など)と座標が必要となる。分子情報は psf ファイルまたは、pdb ファイルから読み取ることができる。座標は dcd ファイルまたは、pdb ファイルから読み取ることができる。ファイルの読み込みは vmd を立ち上げ、Main 画面から「File」タブを選び、「New Molecules」を選択することで出力される「Molecule File Browser」にてファイル名、ファイルの種類を選択する(図8)。また、dcd ファイルには原子や残基の種類や名前などの情報は含まれていないため、psf/pdb などの原子や残基の種類を持つファイルを先に読み込み、その後 dcd ファイルを読み込む必要がある。
工夫とコツ
温度と圧力の調整について
MD の予備計算において、温度と圧力の調整段階は、非常に重要な部分である。ターゲットとなるシステムにより適宜変更することが必要な場合もあるが、基本としては、
- 体積一定(NVT)計算で温度をゆっくり上昇させ、標的温度で十分に安定になっていることを確認した後に、圧力一定(NPT)計算で体積の調整を行う。温度上昇のシミュレーションでは GENESIS の Annealing オプションを使うのも良い。
- 本計算が NVT 条件であったとしても、シミュレーションボックス内に真空のバブルができるのを防ぐため、平衡化の前に必ず 1~2 ns 程度の NPT 計算を行う。
- 時間幅は 2 fs 以下にする。
- Multiple time step 法(r-RESPA)は使わない。
- 初期構造が極端に不安定な構造の場合は、時間幅を 0.5~1 fs、NVT 条件、温度を低温(100K 以下)、contact_check オプションを YES にして計算を行う。複数回計算を行い、冒頭の contact_check オプションによる不安定な結合の表示が出力されなくなるまで行う。
- E で記した設定でもエネルギー値が安定にならない場合は、以下の可能性が高い。
(ア) セットアップが正しく行われないためタンパク質分子モデルの構造が正しくない。次項を参考にされたい。
(イ) GENESIS のコントロールファイルでのパラメータが設定されていない。高速化スキームや力場の必要条件から、利用者がコントロールファイルに設定したパラメータと、実際に計算で用いられるパラメータが一致しない場合がある。ログファイルの STEP 3 で確認する。
タンパク質分子モデルの確認時の注意
タンパク質分子モデルの構造を見直すときには、タンパク質の二次構造、高次構造が不自然になっていない事を確認する。更にタンパク質、溶媒の双方がシミュレーションボックスに正しく配置されているかを確認する必要がある。可視化ソフトでタンパク質分子モデル全体を表示。水分子で作られたボックスの内部に水溶性タンパク質が置かれているような状態になっていることを確認。タンパク質、脂質膜分子に対しては、同一分子が同じシミュレーションボックスにある事も重要な確認事項である。周期境界条件計算の場合は、計算が進んでいくうちに、溶媒分子(水、脂質膜分子)はシミュレーションボックスより見かけ上広がるように見える(図9)。これは誤りではない(周期境界条件については [5] を参考にされたい)。しかし、タンパク質分子モデルの形状(真空のバブルができていないか、シミュレーションボックスよりタンパク質が広がっていないか)の確認のために、必ず分子をボックス内に再配置してから確認する。
GENESIS が異常終了する場合の対処法
- エラーメッセージが “SHAKE algorithm failed to converge: ” であるほとんどの場合は、分子内拘束モジュールのエラーではなく、分子構造が壊れ、水素を含む共有結合長が正常範囲に収まらなくなることが原因である。以下のステップで構造を確認する。
- 出力から各エネルギー値や温度が異常に上がっていないかを確認
- エラー直前のステップ数でトラジェクトリを出力させ、構造を可視化ソフトで目視
- 構造最適化、温度上昇シミュレーションで利用した contact_check オプションを使い、不安定な構造がないかを確認 構造が壊れる原因としては、最適化、温度・体積調整計算の不十分さ、コントロールファイルのパラメータの誤りなどが考えられる。パラメータの誤りがない場合は、「温度と圧力の調整について」の (E) を参考にし、最適化からもう一度行う事を推奨する。
- エラーメッセージが “Memory allocation error” である、もしくはエラーメッセージが表示されない何らかの原因でタンパク質分子モデルが破壊され、正しくない計算が起きている事が疑われる。1と同様に構造が安定になっている事を確認する。GENESIS では、確認用に原子数、シミュレーションボックスサイズを表示させ、配列外アクセスなどのソフトウェアの問題を検知しながらシミュレーションを行うデバッグオプションが搭載されている(ただし、実行時間は通常の5倍程度かかる)。デバッグオプションのコンパイルの仕方は以下である
$ ./configure --enable-debug=3 $ make clean $ make ‒j 4 install
文献
- Jung, J.*, Mori, T.* et al. (*equally contributed), WIREs Comput. Mol. Sci., 5, 310–323 (2015)
- Kobayashi, C.*, Jung, J.* et al. (*equally contributed), J. Comput. Chem., 38, 2193–2206 (2017)
- Visual Molecular Dynamics: http://www.ks.uiuc.edu/Research/vmd/
- Jo, S., et al., J. Comput. Chem., 29, 1859–1865 (2008)
- 上田顯, 分子シミュレーション―古典系から量子系手法まで―, 裳華房 (2003)
-
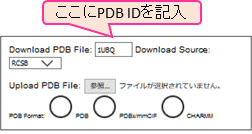
図1:CHARMM-GUI の PDB ID の指定画面 -
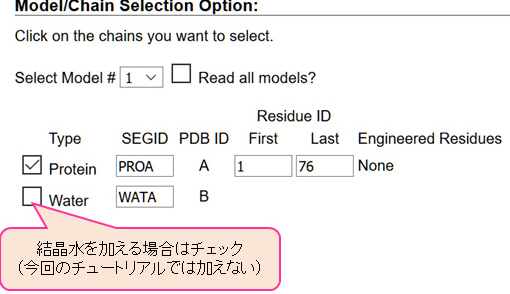
図2:CHARMM-GUI のPDB 中の分子の確認 -

図3:CHARMM-GUI の水分子、イオンの追加画面 -
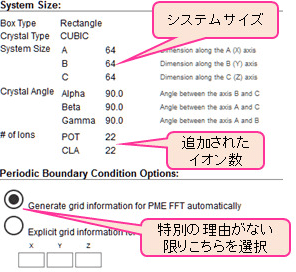
図4:CHARMM-GUI で作成した分子モデルの確認画面 -
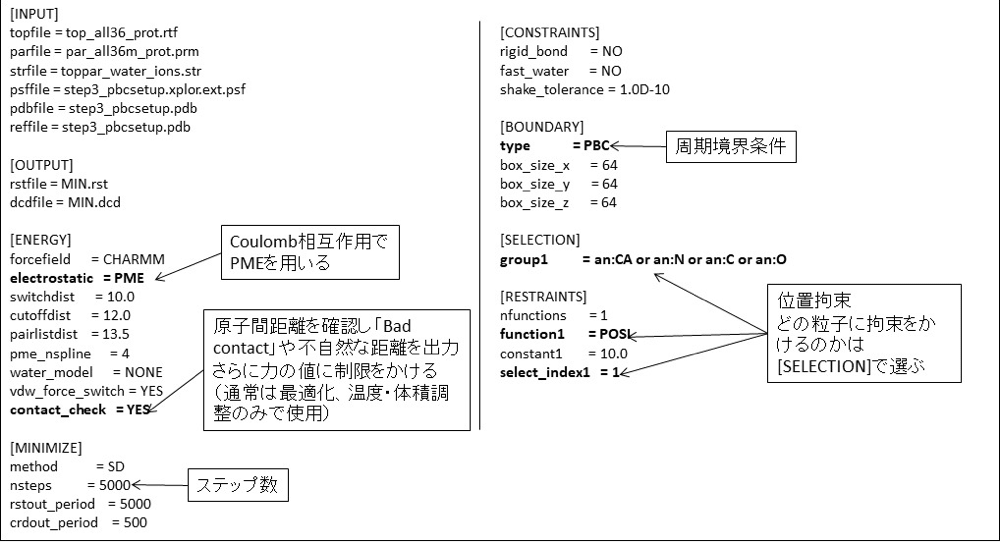
図5:構造最適化計算のためのコントロールファイル(全体) -

図6:温度上昇シミュレーションのコントロールファイルの抜粋 -
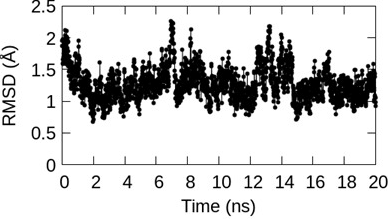
図7:本計算における解析の一例。初期座標からの RMSD -
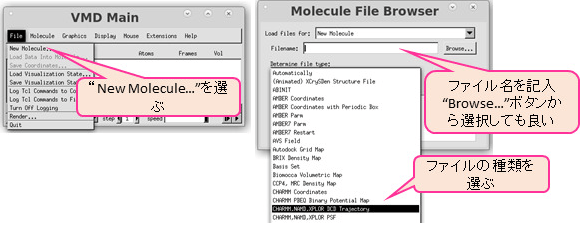
図8:VMD によるトラジェクトリ(dcd ファイル)の表示。左は VMD Main 画面。右はファイル選択画面。 -

図9:VMD で表示した周期境界条件での蛋白質分子モデル。青い箱はシミュレーションボックスを表す。左はトラジェクトリファイルをそのまま表示したもの。右は VMD の pbctool を使いボックス内に分子を再配置したもの。
概要
分子動力学法(Molecular Dynamics, MD)は、原子・分子間に働く力を用いて運動方程式を数値的に解くことで、粒子の運動を求める方法である。揺らぎや運動などのダイナミクス解析、自由エネルギー面の探索などに利用されている。近年は、計算機と計算手法の発展により、計算化学の専門家だけでなく実験研究者にもモデリング、リファインメント、創薬・分子デザインに広く利用されるようになっている。本稿では、理化学研究所計算科学研究センター粒子系生物物理研究チームを中心に開発されている MD ソフトウェア GENESIS(フリーソフトとして公開中)を用いて、水溶性タンパク質の MD 計算を行う方法を述べる。具体的例を挙げ、GENESIS のインストール、計算するタンパク質分子モデルの準備、MD 計算(予備計算と本計算)を概説する。
イントロダクション
分子動力学法(Molecular Dynamics, MD)は、原子・分子間に働く力(相互作用力)を用いて運動方程式を解くことで、粒子の運動を求める方法である。相互作用する多粒子の運動方程式は数値解法と必要とする。相互作用力の計算には、タンパク質、核酸などの生体高分子を対象とする「力場(force field)」と呼ばれる経験的な関数を用いる。力場による相互作用力計算と運動方程式による時間発展計算には、コンピュータの高い演算能力が大いに威力を発揮する。近年の計算機の発展(特にグラフィックカード(Graphics Processing Unit, GPU)による高速演算)と様々な手法の開発により、計算可能なシステムサイズ、時間オーダーが飛躍的に上がっている。開発された手法の多くは様々な MD ソフトウェアに導入され、利用可能となっている。本稿では、その一つである理化学研究所、計算科学研究センター、粒子系生物物理研究チームを中心に開発されている GENESIS [1,2] を用いて水溶性タンパク質のMD 計算を行う方法を概説する。
装置・ソフトウェア
GENESIS によるシミュレーションの実行には、ソフトウェア、計算機(ワークステーション)、ソフトウェアを動作させるための開発環境が必要となる。近年、多くの大学、研究所の計算機センターではトライアルユースなどが安価(もしくは無料)で提供されている。GENESIS に必要な開発環境は、筆者が知る限り多くの計算機センターで標準的に導入されているものである。また、自然科学研究機構岡崎共通研究施設計算科学研究センターのスーパーコンピュータでは GENESIS が導入されている。計算機のメンテナンスコストを考えると、計算機センターの活用も推奨したい。更に結果を確認するための可視化ソフトウェアが必要となる。
- GENESIS(フリーソフトとして公開中)
最新版は GENESIS のウェブサイトから取得可能。 - 本稿で紹介する入力ファイル一式は、ここから入手可能である。ファイルを以下のように展開する。
$ tar xvhf Protocol_ howtoGENESIS.tar.bz2 $ cd Protocol_ howtoGENESIS/Protocol_ howtoGENESIS/には以下のディレクトリが存在する。1_minimization/ 2_A_heating/ 2_B_NVT/ 2_C_NPT/ 3_equilibration/ 4_production/ molecule_files/ toppar/ - 可視化ソフトウェア: 分子の構造、運動(トラジェクトリ)を確認する。Illinois 大学 Urbana-Champaign 校で開発されている VMD [3] の利用を推奨する。このソフトウェアは、GENESIS や他の MD ソフトウェアで利用されるファイル形式(dcd)を直接読む事が可能。VMD は Linux/Windows/MacOS X にも対応
自分で計算機環境を整える場合は以下が必要となる。
- ワークステーション
- CPU: Intel, SPARC64(HPC-ACE), AMD(Opteron)が動作確認済
速度上から Intel Xeon SandyBridge 世代以降を推奨 - メモリ: 20万原子(注)の計算を1ノードで行うにはソフトウェアに 10 GB 程度の割り当てが必要となるため、1ノード 32 GB 以上が望ましい
- CUDA 対応の GPU カード(NVIDIA)(任意): Compute Capability 3.5, 3.7, 6.0, 6.1, 7.0 が動作確認済
- CPU: Intel, SPARC64(HPC-ACE), AMD(Opteron)が動作確認済
- オペレーションシステム(OS)、開発環境(コンパイラ、ライブラリ)
- Unix 系 OS: Linux, Free BSD, MacOS X などが利用可能
- C、Fortran コンパイラ: gcc, gfortran ならversion 4.4.7 以降、Intel Fortran/C なら version 12 以降
- Message Passing Interface(MPI): OpenMPI(1.10 以降推奨), Intel MPI, 富士通 MPI が動作確認済。OpenMPI は Unix 系OS の多くではパッケージ化されているため、システムツール(apt, dnf/yum, dpkg など)からインストール可能
- Basic Linear Algebra Subprograms(BLAS)、Linear Algebra PACKage(LAPACK)ライブラリ: Unix 系 OS の多くではパッケージ化されているため、システムツールからインストール可能。また、Intel MKL, 富士通 SSL の利用可能
- GNU autoconf: version 1.3 以降
- (GPU 利用の場合のみ)CUDA ライブラリ、コンパイラ: version 8.0 以降
注:分子モデルには溶媒分子も含まれる。20万原子は大体、分子量 60 – 80 k のタンパク質の分子モデルに対応する。ただし、後述のようにタンパク質の形状や性質によりモデルのサイズ(溶媒分子の数)は大きく変化することに注意されたい。
実験手順
- タンパク質分子モデルの準備(セットアップ)
- 構造最適化
- 温度と圧力(体積)の調整
- 平衡化
- 本計算
実験の詳細
GENESIS の内容物
GENESIS は、2つの MD エンジン(spdyn, atdyn)と複数の解析用ソフトウェアからなる。spdyn(SPartial decomposition DYNamics simulation)は超並列計算のために設計され、単精度計算、グラフィックカードの利用が可能である。一方、atdyn(ATomic decomposition DYNamics simulation)は利用者による機能追加が可能なように平易な内部構造となっている。CHARMM, AMBER などの全原子モデルを利用する場合は spdyn を推奨する。本稿は spdyn の利用について記載する。
GENESIS のインストール
入手した GENESIS の tar.bz2 ファイルを展開し、以下の要領でインストールする。実行ファイルは genesis ディレクトリ直下に bin/ ディレクトリに作成されるため、特権ユーザ(super user, root)になる必要はない。
$ tar xvhf genesis-1.3.x.tar.bz2
$ cd ./genesis-1.3.x
$ ./configure
$ make ‒j 4 install
bin/ ディレクトリに spdyn, atdyn と解析用のソフトウェアがインストールされる。
GPU を利用する場合は、以下の手順でインストールを行う。
$ tar xvhf genesis-1.3.x.tar.bz2
$ cd ./genesis-1.3.x
$ ./configure --enable-single --enable-gpu
$ make ‒j 4 install
configure はコンパイル時のオプションを与える機能がある。GENESIS は多くの場合自動的に設定を行うが、手動で設定を行う事もできる。configure で利用可能なオプションは以下のコマンドで確認できる。
$ ./configure --help
正しくインストールされたかを確認するために、テストを行う。テストセット(tests-1.3.x.tar.bz2)も GENESIS のウェブサイトから入手可能である。
$ tar xvhf tests-1.3.x.tar.bz2
$ cd tests/regression_test
sh/bash の場合は
$ export OMP_NUM_THREADS=1
csh/tcsh の場合は
$ setenv OMP_NUM_THREADS 1
test.py を用いて以下のように実行する。
./test.py “mpirun -np 8 実行ファイルの絶対パス/実行ファイル”
例えば、/home/user/genesis にインストールされている場合は、以下となる。
$ ./test.py “mpirun -np 8 /home/user/genesis/bin/spdyn”
また、GPU を利用する場合は gpu オプションを付ける
$ ./test.py “mpirun -np 8 /home/user/genesis/bin/spdyn” gpu
上を実行すると、用意された複数のテストセットが順番に実行される。各テストが正常に動作すると以下のように ”Passed” が表示される。
(例)
Checking ./test_common/dna/CUTOFF
Checking diff between ref and test...
Passed (tolerance = 3.00e-05(ene), 3.00e-03(virial))
また、全てのテストセットの最後に以下のように何個のテストが成功したかが記載される。 テストの総数はテストの種類、GENESIS のバージョンや GPU の有無によって変化する。
(例)
Passed 44 / 44
Failed 0 / 44
Aborted 0 / 44
重要な点としては、このテストは MPI 並列数が8で実行することを想定して作成されているため、必ず MPI 並列数は8で行わなければならない。
CHARMM-GUI を使ったタンパク質分子モデルの準備
次にタンパク質分子モデルの準備(セットアップ)を行う。セットアップを行うツールはいくつか公開されており、本稿では Web ブラウザベースでセットアップができる CHARMM-GUI [4] を利用する方法を紹介する。このソフトウェアは Lehigh 大学の Im 教授のグループで開発されており、セットアップを行い MD の実行ファイルに必要な入力ファイルを出力する。以下に、MD のセットアップを行い、GENESIS に必要なファイルを作成する方法を示す。
- CHARMM-GUI のウェブサイトにアクセスする
- 左側のメニューから Input Generator を選ぶ。
- 左側のメニューから Quick MD Simulator を選ぶ。
- 下側の入力ボックス内で、PDB の ID もしくは、PDB のアップロードを選ぶ。本稿ではテストとして RCSB の PDB からユビキチンの PDB(1UBQ)を指定する(図1)。
- PDB の情報を確認する。本稿の例では入れていないが、Water のチェックボックスにチェックを入れると結晶水もシステムに含まれる(図2)。
- 変異の導入、プロトン化状態(Asp, Glu, Lys, His)の変更・設定、リン酸化や蛍光色素の導入など、タンパク質の情報を変更したい場合には、チェックボックスに適宜チェックを入れる。今回は特に設定を行わない。
- 溶媒水分子、イオンの追加。水分子を詰めたボックスにタンパク質とイオンを入れる方法をとる。このボックスのサイズがシステムのサイズとなる。体積に比例して計算コストは増大する(粒子数の1~2乗)。計算時間を抑えるためには、デフォルトであるタンパク質の端からシミュレーションボックスの端まで10 Åの余裕を持たせる設定が妥当である。ただし、周期境界条件で計算を行うため、この値が小さいとボックス越しに自分同士と相互作用してしまうため、十分に注意が必要となる。特に、Fold/Unfold 間の転移などタンパク質の構造が大きく変化する場合は、タンパク質が取りうる最大のサイズを考えてシステムのサイズを決めなければならない。また、GENESIS ではボックスの形は rectangular のみが可能である。導入するイオンの数は、対象によって適切に設定する(本稿の例ではデフォルトである0.15 M KCl を使用)。ただし、長距離相互作用計算で広く用いられている Particle Mesh Ewald(PME)法での計算では、電荷(net charge)は0でなければならない事に留意されたい。そのため、イオン濃度を設定しない場合でも Add neutralizing ions を選定する(図3)。この過程は5–10分ほど掛かる。
- システムの確認画面となる。システムのサイズ、イオンの数などを確認する。下の Periodic Boundary Condition Options に関しては、特別の理由がない限り変更させない(図4)。
- 出力する形式の選択画面となる。GENESIS を選択。
- セットアップは完了となる。右端の “Download” ボタンを押し、charmm-gui.tgz というファイルを保存する。
- 以下の要領で charmm-gui.tgz を展開し、ディレクトリ内へ移動する。
$ tar xvhf charm-gui.tgz $ cd charmm-gui - 展開された
charmm-guiディレクトリには大量のファイルが存在するが本稿では以下のファイルのみを抜き出して利用する。
①力場ファイル: 力場計算に必要な情報を与える。CHARMM 力場では、それぞれの分子の定義を記載した topology(top)ファイル、原子の質量、電荷、原子間の相互作用のパラメータを記載した parameter(par)ファイル、追加パラメータ(top と par の両方)の情報を記載したstream(str)ファイルからなる。CHARMM の力場の場合は、タンパク質、核酸、糖質、脂質膜分子等の分子種毎にファイルが分かれている。toppar/には現在公開されている全分子種用のファイルがある。全てを読みこませても問題はないが、本稿で説明する水溶性タンパク質の計算ではtop_all36_prot.rtf,par_all36m_prot.prm,toppar_water_ions.strのみが必要である。
②protein structure file(psf)ファイル: タンパク質分子モデルの情報(原子数、残基や原子の種類、相互作用の情報)が記載されている。
step3_pbcsetup.xplor.ext.psf
③pdb ファイル: タンパク質分子モデルの構造(座標)情報が記載されている。
step3_pbcsetup.pdb
GENESIS による MD 計算
A. GENESIS のコントロールファイル
GENESIS では、コントロールファイルと呼ばれるファイルを読み、MD 計算の設定を行う。コントロールファイルでは、主に以下のようなセクションに分けられ、各パラメータが設定されている。
- [INPUT] 入力ファイルの情報(力場ファイルなど)
- [OUTPUT] 出力ファイルの情報(トラジェクトリファイルなど)
- [ENERGY] 力場計算の情報(力場の種類や静電相互作用の計算手法など)
- [DYNAMICS] 時間幅や数値積分の方法の情報
- [MINIMIZE] 構造最適化計算の情報(構造最適化計算のみ利用)
- [CONSTRAINT] 分子内拘束(SHAKE/RATTLE, SETTLE)の情報
- [ENSEMBLE] アンサンブル計算の情報(温度や圧力など)
- [BOUNDARY] シミュレーションボックスの情報(サイズや周期境界条件など)
- [RESTRAINTS] 拘束(restraint)ポテンシャルの情報
- [SELECTION] 拘束計算などに利用する原子・分子グループの情報
また、spdyn/atdyn などの MD プログラム、解析プログラムを以下のように実行することでコントロールファイルのテンプレートが作成される。
$ 実行ファイルのパス/実行ファイル ‒h ctrl > コントロールファイルのテンプレート
出力されたテンプレートは、vim や emacs などのエディターを使い修正し、コントロールファイルを作成する。
B. GENESIS の実行方法
GENESIS は以下のように実行される。
mpirun -np 8 実行ファイルのパス/spdyn コントロールファイル > ログファイル
ログファイル内の出力は7段階(STEP 0-6)に分かれており、それぞれ以下の情報が出力される。
- STEP 0: 計算環境(ハードウェア、ソフトウェア)
- STEP 1: コントロールファイルでの入力パラメータ
- STEP 2: 並列数(プロセス、スレッド数)
- STEP 3: 分子・エネルギー関数情報
- STEP 4: 初期座標でのエネルギー値
- STEP 5: 各ステップでの各エネルギー値、温度、体積などのデータ
- STEP 6: 演算時間
STEP 5の各ステップのデータの出力と並んで、STEP 3の分子・エネルギー関数情報の出力は重要である。この出力と、コントロールファイルにて設定したパラメータや分子、力場ファイルが整合しているかを確認されたい。ここに設定した物と異なるパラメータ、ファイルが出力されている場合は、意図しない計算を行う場合がある。その場合は、出力内の警告(WARNING)を参考に適宜修正を行う。
C. GENESIS によるMD 計算の流れ
MD の計算は、一般的には以下の手順で実行される。
- 構造最適化
- 温度と圧力(体積)の調整
- 平衡化
- 本計算
手順1–3がタンパク質分子モデルを温度、圧力などの計算条件に合わせる予備計算である。人為的な手法で作られた初期構造は、原子間の距離が近づき過ぎ、不自然な原子配置を持つことが少なくない。不自然な原子配置では、特定の原子間の相互作用力が大きくなり、数値解の誤差が増大し、粒子の運動を正しく求められない。その場合は、分子の一部のみが大きく動き、最悪の場合はタンパク質分子モデル全体が崩壊することもある。このような「不安定なシミュレーション」を避けるため、予備計算を慎重かつ十分に行う事が必要である。
CHARMM-GUI から出力されるコントロールファイルはミニマルな仕様となっているため、本稿では著者が作成したファイルを例に説明を行う。
1. 構造最適化(ディレクトリ 1_minimization/)
構造最適化は予備計算の第一段階として、相互作用エネルギー値が下がる方向に粒子を動かし、不安定な原子配置を取り除く計算である。GENESIS では現在、最急降下法(Steepest decent method)が利用可能である。図5に GENESIS のコントロールファイルを示す。
この段階では不安定な原子配置が存在するため、計算のターゲットになるタンパク質・高分子・リガンドには位置や距離に拘束を適宜導入して、極端に分子が動かないようにすることが必要である。ステップ数は、あくまで極端にエネルギーが高い構造をある程度解消することが目的であるため、数千ステップ程度で良い。GENESIS では、不自然な距離にある原子対を出力し、かつ、相互作用力の値に制限をかける “contact_check” というオプションが搭載されている。構造最適化や温度・体積を調整するシミュレーションでは利用されたい。数千ステップ後でも、エネルギー値が上下するなど落ち着かない場合は、「工夫とコツ」(後述)の「タンパク質分子モデルの確認時の注意」を参照し、構造の確認を行う。
2. 温度と圧力(体積)の調整
システムを計算する標的温度、圧力に慣らす。安定した本計算を行うためには最も重要なステップである。温度上昇、温度一定、体積調整の3段階の計算を行う。
A) 温度上昇シミュレーション(ディレクトリ 2_A_heating/)
温度上昇シミュレーションは、構造最適化によりある程度安定化された構造に対して、システムの温度をゆっくりと上昇させ、標的温度に至らせるものである。GENESIS では、“Annealing” オプションを使うことが可能である(図6)。少しずつ標的温度を上昇させながら複数回シミュレーションを行っても良い。
B) 温度一定シミュレーション(ディレクトリ 2_B_NVT/)
前項での温度上昇シミュレーションでは、標的温度での計算は500 step しかできず、システムの温度の調整としては不十分である。そのため、体積調整シミュレーションを行う前に温度一定(NVT)のシミュレーションを行う。この段階では、ログファイルから温度の時間変化を確認し、設定した温度の周辺を揺らいでいることを確認する。体積が変化しないため、シミュレーションボックス内に真空のバブルができることがある。これは、人為的に作成したシミュレーションボックスは、分子の密度が必ずしも正しくないためである。このバブルは次のステップで解消させる。
C) 体積(密度)調整シミュレーション(ディレクトリ 2_C_NPT/)
人為的に作成したシミュレーションボックスをシステムに最適な密度に合わせるためにNPT 計算をおこなう。特に本計算がNVT 条件の場合では、温度上昇、温度一定シミュレーションの後に直接平衡化を行うと、シミュレーションボックス内のバブルが解消されない。そのため、平衡化の前に必ず1~2 ns 程度の NPT 計算を行い、最適な密度に合わせる必要がある。体積の時間変化を確認し、体積がある値の周辺で揺らぐ状態にある(ドリフトしない)事を確認する。また、圧力はステップ数毎の揺らぎが大き過ぎるため、この場合の指標としては適切ではない。体積変化により位置拘束の力が急速に上がり、粒子の運動や圧力の計算が正しく行われなくなる恐れがあるため、基本的には位置拘束は使わない。
3. 平衡化(ディレクトリ 3_equilibration/)
本計算で利用する条件と同じ条件でシステムを安定化すべく、同じ条件での計算を行う。これ以降の計算では、contact_check オプションは必ず外すこと。2 fs 以上の時間幅や長距離相互作用計算や温度・圧力制御を数ステップ置きに行う Multiple time step 法などの高速化スキームは、この段階から使用する。最低でも 20 ns 程度の計算が必要となる。これは、20 ns で十分という意味ではない。平衡化に要するステップ 数は計算するシステムによって大きく異なるため、温度、体積(NPT 条件の場合)、ポテンシャルエネルギーの時間変化がドリフトしていない事を確認する。
4. 本計算(ディレクトリ 4_production/)
実際にデータを取るための計算である。サブ μs から、最近では数十 μs オーダーの計算も珍しくなくなった。粒子の軌跡をトラジェクトリとして書き出しを行うことで解析を行い、様々な原子・分子の性質を計算することが可能となる。チュートリアルのコントロールファイルでは例のために 20 ns となっているが、実際の研究ではもっと長時間の計算が必要であることを強調したい。
MD 計算は、極小値に向けて値が収束していく計算と異なり、出力される温度、体積、エネルギー値などの数値データだけではその成否を判断できない。そのため、本計算だけでなく予備計算の段階から、分子たちが意図した条件内で運動している事を、タンパク質の揺らぎ(RMSF)、初期座標からの RMSD、リガンド、残基間の塩橋や水素結合ネットワークの構造情報を随時計測し、その時間変化を丁寧に追うことも必要となる(図7)。
D. 可視化ソフト(VMD)による MD 計算のトラジェクトリ表示
VMD を用いてトラジェクトリを可視化するためには、分子情報(原子や残基の名前など)と座標が必要となる。分子情報は psf ファイルまたは、pdb ファイルから読み取ることができる。座標は dcd ファイルまたは、pdb ファイルから読み取ることができる。ファイルの読み込みは vmd を立ち上げ、Main 画面から「File」タブを選び、「New Molecules」を選択することで出力される「Molecule File Browser」にてファイル名、ファイルの種類を選択する(図8)。また、dcd ファイルには原子や残基の種類や名前などの情報は含まれていないため、psf/pdb などの原子や残基の種類を持つファイルを先に読み込み、その後 dcd ファイルを読み込む必要がある。
工夫とコツ
温度と圧力の調整について
MD の予備計算において、温度と圧力の調整段階は、非常に重要な部分である。ターゲットとなるシステムにより適宜変更することが必要な場合もあるが、基本としては、
- 体積一定(NVT)計算で温度をゆっくり上昇させ、標的温度で十分に安定になっていることを確認した後に、圧力一定(NPT)計算で体積の調整を行う。温度上昇のシミュレーションでは GENESIS の Annealing オプションを使うのも良い。
- 本計算が NVT 条件であったとしても、シミュレーションボックス内に真空のバブルができるのを防ぐため、平衡化の前に必ず 1~2 ns 程度の NPT 計算を行う。
- 時間幅は 2 fs 以下にする。
- Multiple time step 法(r-RESPA)は使わない。
- 初期構造が極端に不安定な構造の場合は、時間幅を 0.5~1 fs、NVT 条件、温度を低温(100K 以下)、contact_check オプションを YES にして計算を行う。複数回計算を行い、冒頭の contact_check オプションによる不安定な結合の表示が出力されなくなるまで行う。
- E で記した設定でもエネルギー値が安定にならない場合は、以下の可能性が高い。
(ア) セットアップが正しく行われないためタンパク質分子モデルの構造が正しくない。次項を参考にされたい。
(イ) GENESIS のコントロールファイルでのパラメータが設定されていない。高速化スキームや力場の必要条件から、利用者がコントロールファイルに設定したパラメータと、実際に計算で用いられるパラメータが一致しない場合がある。ログファイルの STEP 3 で確認する。
タンパク質分子モデルの確認時の注意
タンパク質分子モデルの構造を見直すときには、タンパク質の二次構造、高次構造が不自然になっていない事を確認する。更にタンパク質、溶媒の双方がシミュレーションボックスに正しく配置されているかを確認する必要がある。可視化ソフトでタンパク質分子モデル全体を表示。水分子で作られたボックスの内部に水溶性タンパク質が置かれているような状態になっていることを確認。タンパク質、脂質膜分子に対しては、同一分子が同じシミュレーションボックスにある事も重要な確認事項である。周期境界条件計算の場合は、計算が進んでいくうちに、溶媒分子(水、脂質膜分子)はシミュレーションボックスより見かけ上広がるように見える(図9)。これは誤りではない(周期境界条件については [5] を参考にされたい)。しかし、タンパク質分子モデルの形状(真空のバブルができていないか、シミュレーションボックスよりタンパク質が広がっていないか)の確認のために、必ず分子をボックス内に再配置してから確認する。
GENESIS が異常終了する場合の対処法
- エラーメッセージが “SHAKE algorithm failed to converge: ” であるほとんどの場合は、分子内拘束モジュールのエラーではなく、分子構造が壊れ、水素を含む共有結合長が正常範囲に収まらなくなることが原因である。以下のステップで構造を確認する。
- 出力から各エネルギー値や温度が異常に上がっていないかを確認
- エラー直前のステップ数でトラジェクトリを出力させ、構造を可視化ソフトで目視
- 構造最適化、温度上昇シミュレーションで利用した contact_check オプションを使い、不安定な構造がないかを確認 構造が壊れる原因としては、最適化、温度・体積調整計算の不十分さ、コントロールファイルのパラメータの誤りなどが考えられる。パラメータの誤りがない場合は、「温度と圧力の調整について」の (E) を参考にし、最適化からもう一度行う事を推奨する。
- エラーメッセージが “Memory allocation error” である、もしくはエラーメッセージが表示されない何らかの原因でタンパク質分子モデルが破壊され、正しくない計算が起きている事が疑われる。1と同様に構造が安定になっている事を確認する。GENESIS では、確認用に原子数、シミュレーションボックスサイズを表示させ、配列外アクセスなどのソフトウェアの問題を検知しながらシミュレーションを行うデバッグオプションが搭載されている(ただし、実行時間は通常の5倍程度かかる)。デバッグオプションのコンパイルの仕方は以下である
$ ./configure --enable-debug=3 $ make clean $ make ‒j 4 install
文献
- Jung, J.*, Mori, T.* et al. (*equally contributed), WIREs Comput. Mol. Sci., 5, 310–323 (2015)
- Kobayashi, C.*, Jung, J.* et al. (*equally contributed), J. Comput. Chem., 38, 2193–2206 (2017)
- Visual Molecular Dynamics: http://www.ks.uiuc.edu/Research/vmd/
- Jo, S., et al., J. Comput. Chem., 29, 1859–1865 (2008)
- 上田顯, 分子シミュレーション―古典系から量子系手法まで―, 裳華房 (2003)
