

概要
本プロトコールは、セレン化や重原子置換を行わない非標識天然型タンパク質のままで、構造未知の新規タンパク質のX線結晶構造解析を行う手順、及び留意点をまとめたものである.タンパク質の結晶構造解析を行う場合、構造因子の位相角を計算するために、結晶にPd、Pt、Au、Hg、ランタノイドといった重原子化合物を直接導入するか、遺伝子工学的手法によって、セレンをタンパク質分子に導入する必要があった。もともとタンパク質が持っているメチオニン及びシステインのイオウ原子の異常散乱を利用できれば、こういった改変をすることなく、位相計算を行うことが可能になる。しかし、イオウ原子の異常散乱の全回折強度に対する寄与は、重原子やセレンと比較して非常に小さく、容易に測定誤差に埋もれてしまう。イオウ原子の異常散乱は、X 線結晶構造解析で使用される波長域では波長に依存して長波長ほど大きくなるため、長波長の X 線を用いた測定が有効である。しかし、長波長の X 線は物質による吸収が大きいため、測定データの精度が低下する。これを回避するには測定の工夫が必要となり、さらにデータ解析の際にも、注意すべき点がある。
装置・器具・試薬
タンパク質結晶、長波長が使用できるビームラインや実験室系のX線源(1.5 – 2.3Å 程度)、試料低温吹付装置、タンパク質結晶凍結器具(Hampton 製ナイロンループ、CrystalCap 等)、二次元 X 線検出器
実験手順
1)サンプルの評価
2)結晶のマウント
3)X線回折データ収集
4)データ解析
実験の詳細
1)サンプルの評価
試行するサンプルが、非標識天然型タンパク質のままでの位相決定(Native-SAD)に適しているかどうかの指標の1つに、イオウの含有率が挙げられる。タンパク質に含まれるイオウ原子は、メチオニン及びシステイン残基に由来するが、通常のタンパク質であれば、これらの数は多ければ多いほど、位相決定には有利に働く。異常散乱の虚数項f“は、原子種及び波長に依存し、このf“に由来する構造因子の異常散乱差ΔFの構造因子Fに対する比<ΔF>/<F>(1,2)によって、位相決定の可否を、ある程度推測することができる。これはタンパク質分子の大きさに対するイオウ原子の数によるが、波長にも依存する。<ΔF>/<F>は http://castor.sci.hokudai.ac.jp/watanabe/S-count/ のようなツールを使うと、アミノ酸配列からも見積もることができる。当研究室の実績から、この値が1.0%以上であると、位相計算が成功する可能性が高い。あらかじめ<ΔF>/<F>値を計算し、それが1.0%程度以上あれば、まずNative-SADによる位相決定を試行するとよい。およその目安として、1.5ÅのX線を使用する場合、100残基あたりイオウ5個、2.3ÅのX線では2個程度あれば良い。
2) 結晶のマウント
長波長X線は、物質との相互作用が大きい。そのため、結晶周辺に存在する結晶以外の物質による吸収効果が回折データの精度に大きく影響してしまう。結晶以外の物質とは、具体的にはクライオ溶液である。この影響を最小限に抑えるために、タンパク質結晶のマウント方法を工夫しなくてはならない。
・通常のマウントループをそのまま使用する場合
通常のマウントループを用いてタンパク質結晶をマウントする場合は、結晶のサイズとほぼ同じ大きさのループを使用し、結晶とループの間の溶液が極力少なくなるようにする。
・通常のマウントループを加工して使用する場合
通常のマウントループを加工することで、溶液量を極端に少なくして、タンパク質結晶をマウントすることが可能である。
・釣り針型(図1)
ナイロンループの一部をカットすることで、釣り針状のマウント治具を作ることができる。
・先割れ型(図2)
ループ中央付近を、斜めにカットすると、先が二股に分かれた治具を作ることができる。使用する際には、その二股の間にタンパク質結晶を挟む。
これら2つの方法は、通常のマウントループを使用する場合と比較して、結晶周辺の溶液の体積を減らすことができる。
また、プロテインウェーブ社(http://www.pro-wave.co.jp/)のリソループや、創晶社(http://www.so-sho.jp/)のクリスタルキャプチャー、MiTeGen社(http://www.mitegen.com/:日本代理店は、ファルマアクセス社http://www.pharmaxess.com/)のMicroMountなどの市販ツールを使うことで、通常のマウントループを使うよりも結晶周囲の溶液量を減らして凍結支持できる。ただし、ここで紹介した方法や、これらの市販のツールを使用する場合は結晶が乾いてダメージを受けやすいので注意が必要である。
・キャピラリートップマウントの利用(図3)
タンパク質結晶にダメージを与えずに、なおかつ、結晶の周囲にクライオ溶液がほぼ存在しない状態で結晶をマウントする方法が開発されている(3)。この方法は、極細のガラスキャピラリーをマウントループの軸に用い、結晶を凍結させる寸前に、ガラスキャピラリーを通じてクライオ溶液を吸引除去する。そのため、ガラスキャピラリーとその治具を製作し、キャピラリーの先端に、ナイロンループを接着しなければならない。使用する先細のガラスキャピラリーは、細胞生物学に使用されているキャピラリープラー等を使用して製作することが出来る。興味のある方は筆者らに連絡していただけば方法を紹介することができる。
(注:この方法は以前はループレスマウントと呼んでいたが、ループは残してもデータ精度には大きく影響しないので残してしまう場合があり、そのため最近は「キャピラリートップマウント」と呼んでみている。)
なお、クライオ溶液として、パラトンやパラフィンオイルといった油脂溶液を用いる例が見られる。本手法についても、こうした油脂溶液の使用は問題ないが、いずれにしても溶液量が少ないほうが良いことに変わりはないので、できる限り取り除いた状態で凍結、支持すると良い。パラトンは粘性が高いため、結晶周囲の溶液を取り除きにくく、この方法には不向きである。筆者らは適当な粘度のフォンブリンオイルを使っている。
3) X線回折データ収集
イオウ原子の微弱な異常散乱差を測定するために、比較的長い波長のX線を用いて回折実験を行う。放射光施設では1.7、 1.8、 2.0 Åといった波長が用いられる。前述のように、異常散乱寄与の虚数項f”は、イオウ原子に関しては、長波長ほど値が大きくなるが、波長が長くなるにしたがって、データ精度の低下も顕著になる。実験室系のX線発生装置では、 Cu Kα線(1.54 Å)以外に、Cr Kα線(2.29Å)が用いられる。長波長X線による回折データ測定では、空気や検出器の窓材によるX線吸収の影響も小さく抑える必要がある。実験室系のX線発生装置でCr Kα線を使用する場合は、ヘリウムパスや、検出器前面の黒紙を炭素含有の高分子シートと交換した装置が市販されている(4)。さらに、コリメータの先端に、キャップ状の金具を付加し、コリメータから結晶までの空気による散乱を減少させることもできる(3)。データ測定に関しては、一般に、通常の回折データ測定よりも冗長度(Redundancy)を上げて統計精度を上げると、良い結果が得られる。しかし、露光時間が長くなると放射線損傷による回折データ精度の低下が顕著に表れることもある。また、どの程度冗長度を上げると解析可能なデータが得られるのかというような明確な指標は存在しない。当研究室の実績では、放射線損傷による回折強度の顕著な減衰など、明確な問題が存在しない場合は、冗長度が15~20以上であれば位相決定に成功し、解釈可能な電子密度が得られている。360度の範囲の測定データの冗長度は、結晶の属するラウエ群の等価位置の数×2 (ここで2倍は反射球に入るときと出るときの2回)で大まかに計算できる。例えば、P212121であれば、ラウエ群はmmmで等価位置は8なので、8×2=16となり、360度振動写真を撮ると、解析に必要な最低限の冗長度はほぼ確保できる計算になる。
4) データ解析
データ解析手法は、通常の単波長異常散乱法(SAD法)と基本的には変わらない。ここではHKL2000(5)、SHELXC/D(6)を用いる例を紹介する。測定した回折イメージを積分、スケーリングする。HKL2000を使用する場合、Scaleタブ中のScale Anomalousオプションに関しては、高冗長度に測定できた場合はOnに、結晶の対称性が低いなどの理由で、高冗長度に測定できなかった場合は通常のAnomalousオプションにしている。一旦データをマージしてしまうと、もとのデータが持っていた統計情報が失われてしまい、この後の処理でSHELXC(7)を使用した場合に統計情報の容易な確認が出来なくなるため、等価反射の強度をマージしないままの回折強度データファイルも作っておいたほうが良い。Scalepackではno merge original indexオプションを付けておくと可能である(図4)。
イオウ原子及びその他の異常散乱原子の位置決定は、SHELXD(8,9)を用いて行う。SHELXCやSHELXDを使用する際に、HKL2MAP(10)を用いるとグラフィカルに操作することができる(図5、 6)。まずSHELXCで異常散乱原子の部分構造に相当する構造因子を計算するが、入力としてマージしていない回折強度データを用いることで、I/σの他、測定データを任意に分割した2つのサブセットで独立に計算した異常散乱差の相関 CC(anom)と、異常散乱シグナルの大きさ<ΔF/σ(F)>という2つの統計値を得ることができ (図7)、測定データの精度を判断することができる。回折強度が精度良く測定できていれば、任意に分割したサブセットで計算した異常散乱差間にばらつきが無いはずであるから相関係数CC(anom)は1に近くなる。また、<ΔF/σ(F)>は、求めた異常散乱差ΔFの構造因子の標準偏差に対する比、すなわちS/N比である。これら2つの値は、分解能が高くなるに従って値が小さくなるが、CC(anom)が低分解能域から50%を割るような場合(通常30%程度以上ある分解能までは使用出来る)や、<ΔF/σ(F)>が1。2 (ノイズレベルは0.8)を割り込むような場合は、良い測定ができていないと考えられるので、先に進まないで測定をやり直した方が良い。データに問題が無い場合には、CC(anom)が30%、<ΔF/σ(F)>が1.5程度までの分解能のデータを使用すると良い。HKL2MAPを使用している場合は、これらを考慮して自動的に分解能を提案してくれる。ただし使用する分解能で結果に差が生じるので、困難なデータの場合、いくらかの試行錯誤は必要となる。
次いでSHELXDで異常散乱原子の位置を決定する。通常は100回程度の試行を行い、規格化構造因子の計算値と実測値の相関係数CC値で正解を判断する。イオウのSAD法の場合、全反射を使ったCC値(CC_all)が30%程度あればおそらく正解である。HKL2MAPを使用している場合、全反射のCC値と弱い反射だけを抽出して計算に用いたCC値(CC_weak)の相関プロットを描かせて見ることができる(図8)。また、決定した異常散乱原子の占有率(Occupancy)をサイト毎にプロットしたグラフと、予想されるイオウの数の関係をみることで正解が得られているかを確認することができる(図9)。ただし、長波長X線を用いた天然型タンパク質結晶回折の位相決定では、タンパク質中にはかなり高い確率で、P、Cl、K、Caといった、イオウ以外の、長波長で有意な異常散乱を示す軽原子が含まれていることが多いため、占有率のプロットでは正解が判断出来ない場合も多い。これらの異常散乱原子も位相決定に利用することができるため、SHELXDが決定したサイトが想定されるイオウ原子数より多くなっていても、それらの座標も位相決定に用いると良い場合が多い。逆に、これらの異常散乱原子を導入することを意図して、KClやCaCl2といった塩を含んだ結晶化条件やイオウを含む硫酸アンモニウムの結晶化条件を選択することは有効である。クライオ溶液にハロゲンイオン(Br-、 I-)を意図的に導入し、タンパク質分子表面に結合したそれらイオンの異常散乱を用いて位相決定を行う方法も報告されている(11)。ただし、本手法の利点である「成長した天然型結晶をそのまま使う」という迅速性との兼ね合いで、導入するかどうかを検討すべきであると思われる。
また、求めた異常散乱原子位置の確からしさはSHELXE (図10)で位相改良を進めてみると判断出来る。正解であれば、それを元にして溶媒平滑化によって位相改良を進めれば、より確かな電子密度マップを与えるはずだし、不正解であれば位相改良がちゃんと進まないからである。正しい解の場合は、図11に示した例のように、20サイクルの位相改良でマップの Contrastが50%を超えるような値となる。
イオウ原子によるSAD法で決まる初期位相の精度はあまり良くない。通常は溶媒領域平滑化等を利用する位相改良が必要である。筆者らはSOLVE(12)でイオウ原子位置を精密化して初期位相を計算し、RESOLVE(13,14)で位相改良を行いながらREFMACで部分構造の精密化を行う反復計算(resolve-build script)によって初期構造モデルを構築している。うまく行かない場合は、OASIS-2004(15)、DM(16)、ARP/wARP(17)を組み合わせることによって、位相改良に成功する場合がある(15,18)。
工夫とコツ
Native-SADによる位相決定の可否は、多波長異常分散法や重原子置換法と比較して、タンパク質結晶の良否に大きく依存する。特に、データの分解能が高いほど位相決定が成功することは確かであり、結晶性が極端に良い結晶のみが、Native-SADに適していると思われがちである。しかし、本プロトコールに示したような工夫で測定誤差を可能な限り抑えた長波長X線測定を行うと、2Åに迫るような分解能を持っていなくとも、位相決定は可能である。筆者らの例では、波長1.8Åで測定した分解能2.6Åのデータで、位相決定に成功した例がある。また、SHELXCの計算結果の統計値から判断して、高角の反射を使わずに、2.8Åや3.0Åといった分解能で切って位相決定をした例もある。低分解能データでの位相決定の可能性に関しては、Yaoらによる報告もある(19)。Native-SADの場合、データの良否はRmergeでは判断が出来ないので、SHELXCの出力するCC(anom)や<ΔF/σ(F)>を見て、結果が良好でなければデータ測定もしくは結晶の調製から検討し直す方が近道である。セレノメチオニルタンパク質を調製して結晶化するにはある程度の時間が必要となるので、その間に並行して、再度Native-SADデータ測定を行うことも可能ではないかと考える。
文献
- Dauter, Z., Dauter, M., de La Fortelle, E., Bricogne, G., and Sheldrick, G. M. Journal of Molecular Biology 289(1), 83-92 (1999)
- Dauter, Z., Dauter, M., and Dodson, E. Acta Crystallogr D Biol Crystallogr 58(Pt 3), 494-506 (2002)
- Kitago, Y., Watanabe, N., and Tanaka, I. Acta Crystallogr D Biol Crystallogr 61(Pt 8), 1013-1021 (2005)
- Yang, C., Pflugrath, J. W., Courville, D. A., Stence, C. N., and Ferrara, J. D. Acta Crystallogr D Biol Crystallogr 59(Pt 11), 1943-1957 (2003)
- Otwinowski, Z., and Minor, W. Methods in enzymology 276(A), 307- 326 (1997)
- Sheldrick, G. M. Acta Crystallogr A 64(Pt 1), 112-122 (2008)
- Sheldrick, G. M. SHELXC., Gottingen University, Germany. (2003)
- Schneider, T. R., and Sheldrick, G. M. Acta Crystallogr D Biol Crystallogr 58(Pt 10 Pt 2), 1772-1779 (2002)
- Sheldrick, G. M., Hauptman, H. A., Weeks, C. M., Miller, M. & Usón, I. International Tables for Crystallography, Vol. F, edited by E. Arnold & M. G. Rossmann, , 333-351 (2001)
- Pape, T., and Schneider, T. R. Journal of Applied Crystallography 37, 843-844 (2004)
- Dauter, Z., Dauter, M., and Rajashankar, K. R. Acta Crystallogr D Biol Crystallogr 56(Pt 2), 232-237 (2000)
- Terwilliger, T. C., and Berendzen, J. Acta Crystallogr D Biol Crystallogr 55(Pt 4), 849-861 (1999)
- Terwilliger, T. C. Acta Crystallogr D Biol Crystallogr 59(Pt 10), 1688-1701 (2003)
- Terwilliger, T. C. Acta Crystallogr D Biol Crystallogr 56(Pt 8), 965-972 (2000)
- Yao, D. Q., Huang, S., Wang, J. W., Gu, Y. X., Zheng, C. D., Fan, H. F., Watanabe, N., and Tanaka, I. Acta Crystallogr D Biol Crystallogr 62(Pt 8), 883-890 (2006)
- Cowtan, K. Acta Crystallogr D Biol Crystallogr 55, 1555-1567 (1999)
- Perrakis, A., Morris, R., and Lamzin, V. S. Nat Struct Biol 6(5), 458-463 (1999)
- Watanabe, N., Kitago, Y., Tanaka, I., Wang, J., Gu, Y., Zheng, C., and Fan, H. Acta Crystallogr D Biol Crystallogr 61(Pt 11), 1533-1540 (2005)
- Yao, D. Q., Li, H., Chen, Q., Gu, Y. X., Zheng, C. D., Lin, Z. J., Fan, H. F., Watanabe, N., and Sha, B. D. Chinese Physics B 17(1), 1-9 (2008)
-
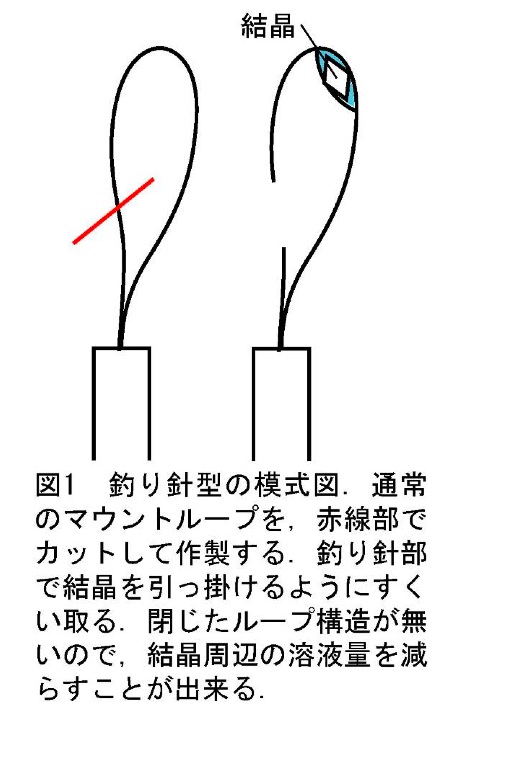
図1: -
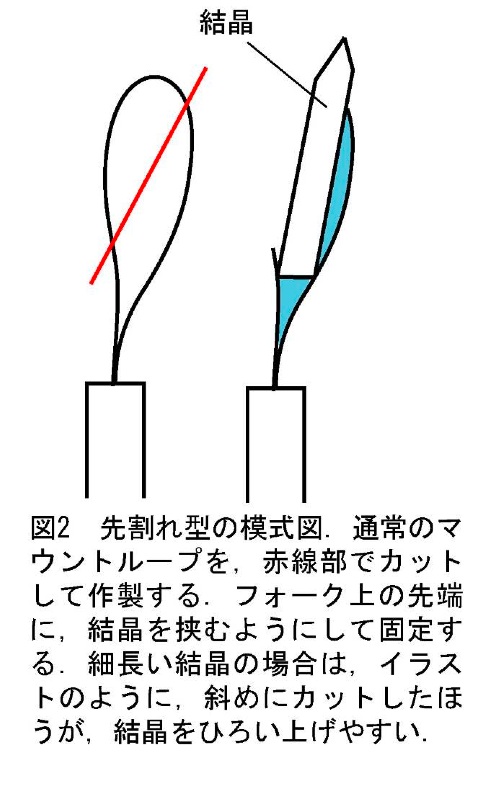
図2: -
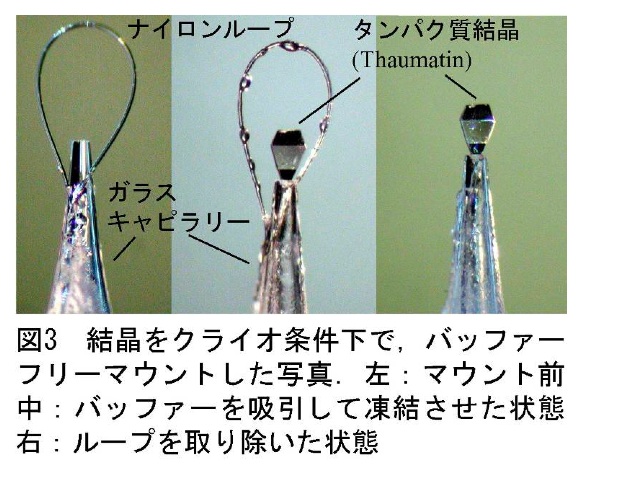
図3: -

図4: -
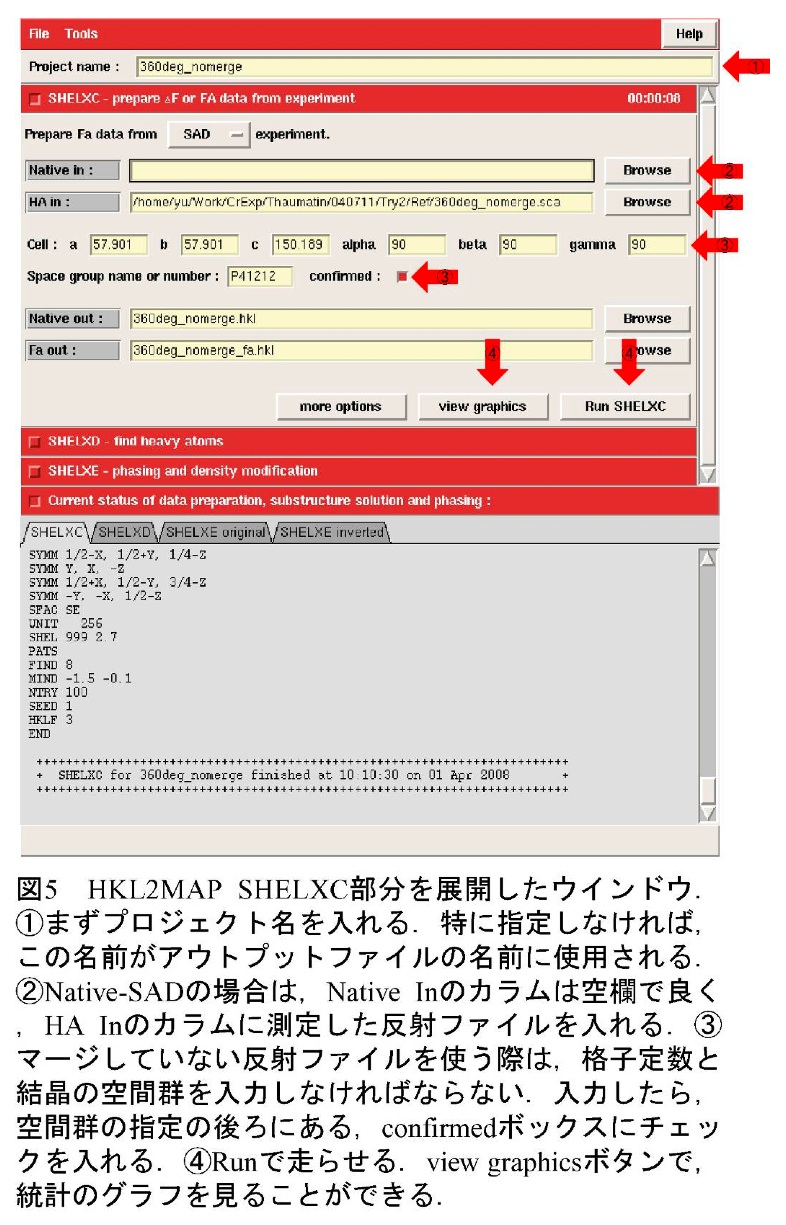
図5: -

図6: -
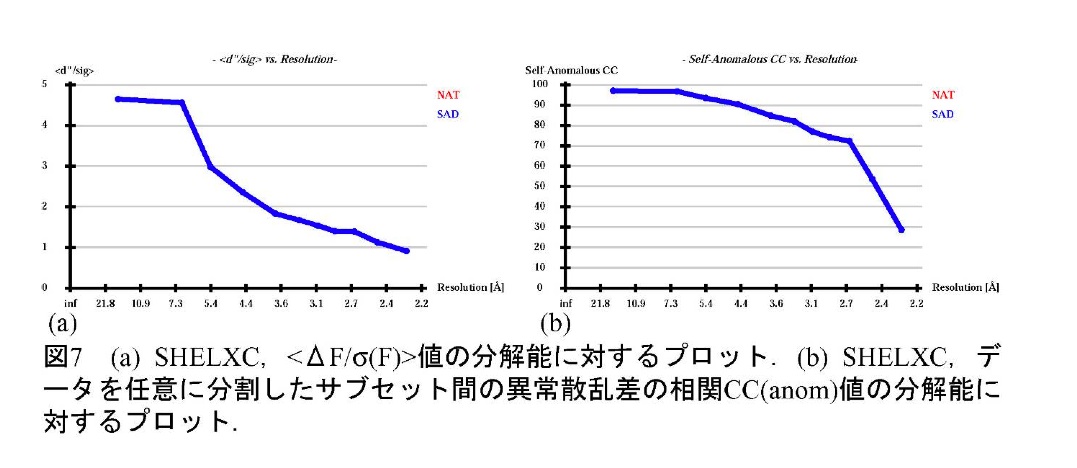
図7: -
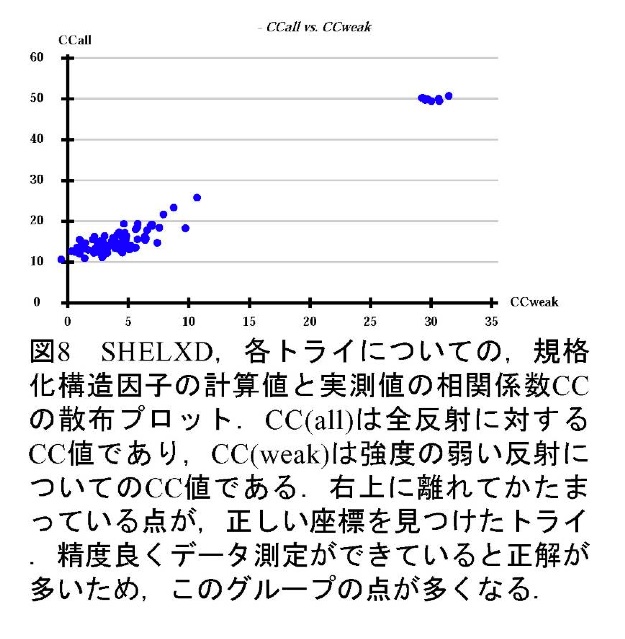
図8: -
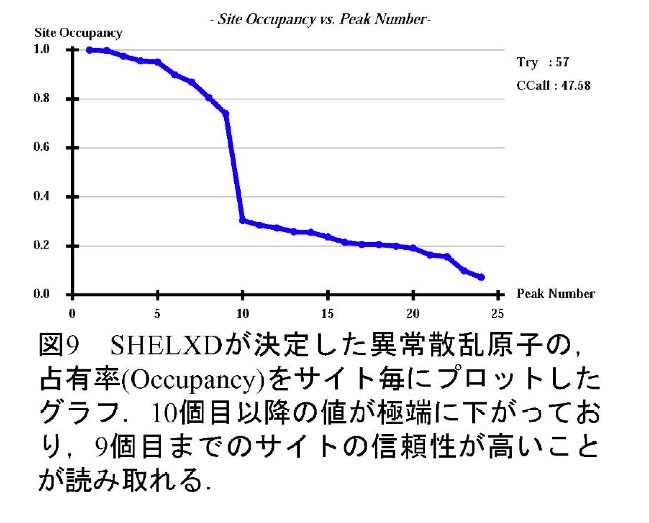
図9: -

図10: -
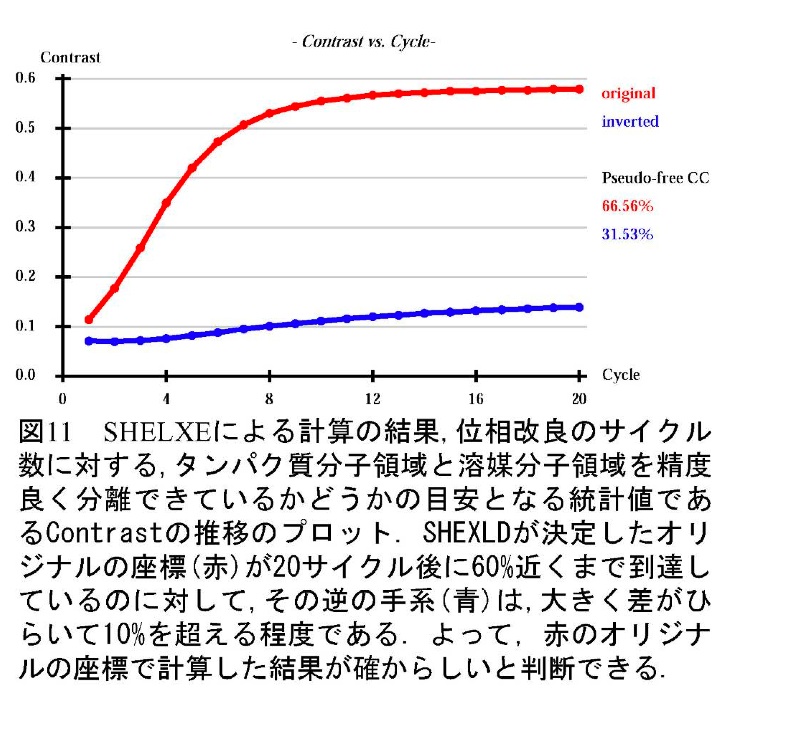
図11:
