

概要
ゲル濾過カラムクロマトグラフィー(以下ゲル濾過と略)はタンパク質や多糖類、核酸などの生体分子を精製する際に用いられる代表的な方法の一つである。ゲル濾過では、カラムに充填された担体にサンプル通すことで様々な大きさの分子を分子量(ここでは溶液中での分子の大きさの意味)に従って分離することができる。担体には小さな孔が開いてあり、小さい分子はその孔に入り込むため、長い間カラム内にとどまることになる。よって、高分子ほど早く溶出され、低分子は溶出されるまでにより時間がかかる。試料をクロマトグラフィー担体に吸着させるイオン交換クロマトグラフィーやアフィニティクロマトグラフィーとは異なり、ゲル濾過ではサンプルを担体に吸着させないため、バッファー組成が分離能に大きな影響を及ぼすことはない。ゲル濾過はタンパク質などの精製だけでなく、相対分子量の測定にも用いることができる。本プロトコールでは、AKTAexplorer 10S に接続した Superdex 200(GE Healthcare)のプレパックカラムを用いたタンパク質の精製法及び分子量の決定法について記述した。Superdex は短い操作時間で高分離能かつ高回収率な溶出ができるため広く使用されており、この担体を使用した多くの学術論文が出版されている。当研究室でも、多くの精製系においてこの担体を用いたゲル濾過を行なっている。どのゲル濾過担体を用いる際においても、高分離な分画を再現性よく得るためにはカラムのパッキング状態が重要なポイントとなる。また、操作も非常に容易であるのでプレパックカラムを用いたプロトコールについて記述した。
HiLoad 26/60 Superdex 200 pg を用いたタンパク質の精製法
ゲル濾過の主な実験の流れは、カラムの平衡化→サンプルの添加→サンプルの溶出→カラムの洗浄である。タンパク質を精製する際だけでなく、分子量を測定する際もこの基本的な作業手順は同様である。用いるカラムのサイズや流速に依存するが、実験日数はおよそ1日~2日である。まず、始めるにあたり、どのゲル濾過カラムを用いるかを検討しなければならない。ゲル濾過には、様々なタイプの担体があり、分画範囲も様々である。ゲル濾過では、高分子な位置に溶出される(溶出体積が小さい)ほどピークがシャープになるが分離能が低いため分子量の近い他の分子と混ざりやすくなる。一方、低分子な位置に溶出される(溶出体積が大きい)ほどピークは広がるが分離能は高くなるため、分子量の近い他の分子との分離がされやすくなる。このゲル濾過の原理と、どのような大きさのものと目的タンパク質を分離したいのかの両方を考慮してカラムを選択する。Superdex 75 の分画範囲は \(3 \times 10^{3}\) ~ \(7 \times 10^{4}\) であり、Superdex 200 の分画範囲は \(1 \times 10^{4}\) ~ \(6 \times 10^{5}\) である。Superdex は短い操作時間で高分離能かつ高回収率な溶出ができるため、目的タンパク質がこれらの分画範囲内のものだと思われる場合は、まずこれらのカラムで分離を行なってみるとよい。また、分離したいサンプルが大容量の場合(1~13 ml 程度)には、一度に添加可能なサンプル量が多い Superdex 75 prep grade (pg) もしくはSuperdex 200 pg を用いるとよい。
以下に代表例として HiLoad 26/60 Superdex 200 pg を用いたタンパク質精製の実験手順を記述する。
装置・器具・試薬
- HiLoad 26/60 Superdex 200 pg (GE Healthcare)
直径 2.6 cm × 高さ 60 cm - AKTAexplorer 10S(GE Healthcare)
- タンパク質低吸着シリンジフィルター
(例)MILLEX-GV Syringe Driven Filter Unit フィルター材質:親水性 PVDF フィルター孔径:0.22 μm フィルター直径:33 mm(MILLIPORE) - バッファー用メンブレンフィルターユニット
(例)Vaccuum Driven Disposable Filtration System フィルター孔径:0.22 μm 容量:1000 ml(IWAKI)
1)ランニングバッファーの準備
AKTAexplorer を用いた実験では共通していえることだが、用いるものすべてをフィルターにかけて小さな埃などを除いておいたほうがよい。AKTAexplorer を用いた解析は非常に流路が狭く高圧下で行なうため、このような埃が AKTAexplorer 内のフィルターやカラムトップのフィルターを詰まらせ圧を上昇させる原因となる。そこでまず、ランニングバッファーとして用いるバッファーを 0.22 μm のフィルターにかける。さらに気泡が流路に流れ込むと解析の波形を大きく歪ませるので、バッファーを脱気する必要がある。脱気は丁寧に行なうと時間がかかるため、われわれの研究室ではバキュームポンプを用いてフィルターをかけた後にそのまま10分程度吸引し続けることで簡易的な脱気を行なっている。試料となるタンパク質の安定性を考慮してゲル濾過を4℃の冷却状態で行なうため、バッファーを冷却しておく。
ランニングバッファーの一例
- 20 mM Potassium phosphate(pH 8.0)
- 1 M NaCl1
- 10% glycerol
- 5 mM 2-mercaptoethanol
2)カラムの平衡化
冷却したバッファーを温めることなくカラムに流す。この際の流速は、限界圧の 0.3 MPa を超えなければ 4.4 ml/min まで流速をあげても問題ない。しかし、実際に 1 ml/min 以上ではほとんど流したことはない。280 nm での吸光度の測定値が安定し、pH 及び塩濃度がランニングバッファーと等しくなるまでバッファーを流し、カラムを平衡化する(1.2 CV~1.5 CV2のバッファーを流している)。平衡化には流速 1 ml/min だった場合、約6時間半かかることになる。よって実際にサンプルを添加する前日に平衡化を行なっておくとよい。
3)サンプルの添加
使用する担体にも依存するが、ベッド体積の0.5~4%が添加量の目安である。よりピーク分離を高めるためにはサンプル量を2%以下に抑えるとよいが、0.5%以下にしても分離能はそれ以上改善されない。サンプルを濃縮すると、一度の精製での処理容量を上げることができるが、あまりに濃くしすぎると(サンプルの凝集のしやすさにもよるがおよそ 70 mg/ml 以上になると)サンプルの粘性が増し、きれいな分離ができなくなることがある。これらのことを考慮して添加するサンプル量を決め、添加するサンプルをフィルターにかける(フィルターにかけることができないようなサンプルの場合は十分遠心して沈殿物などを除く)。HiLoad 26/60 Superdex 200 pg では、サンプルの添加量は 13 ml 以下にしたほうがよい。サンプル量が少なく脱気は困難であるので、シリンジに直接フィルターをつけるようなタイプのものでフィルターにかけるだけでよい。フィルターにかけたサンプルを迅速にサンプルループにロードする。その際、気泡を十分に除き、気泡が極力入らないようにロードする。
サンプル量の一例
13 ml
この際、サンプルループは Superloop 50 ml(GE Healthcare)を用いた
4)サンプルの溶出
サンプルをロードした後は、プログラムにより自動的に溶出する。サンプルの溶出は 1.2 CV のバッファーを流して行なっている。その際、ロードしたサンプル量をプログラムに入力する(13 ml 以下)。不純物との分離を再現性よく行なうためには、毎回流速も一定にして行なった方がよい。
流速の一例
0.8 ml/min
5)カラムの洗浄及び保存方法
0.5 M NaOH を 1 CV 流し、非特異的に吸着しているタンパク質の大部分を除去した後に、蒸留水を 1.2 CV 以上流す。流したサンプルがそれほど吸着していない場合には、蒸留水を 1.2 CV 以上流すだけで済ませている。また、続けて解析を行なう際には、蒸留水で洗浄することなく次に用いるランニングバッファーをそのまま送液し平衡化してしまっている。
カラムをしばらく使わない場合には、4 CV の蒸留水で洗浄した後に 4 CV の20%エタノール溶液で置換して保存する。この際、カラム内に気泡が発生するのを防ぐため温度変化がないように保存する。
6)その他のカラム
HiLoad 26/60 Superdex 75 pg を用いた場合は全く同様の方法でよい。Superdex 75 HR10/30 や Superdex 200 HR10/30 を用いた場合でもほとんど同様の方法でよい。ただ、ベッド体積が異なるので、添加できるサンプル量は 250 μl までであることに注意が必要である。
Superdex 200 HR10/30 を用いたタンパク質の分子量測定
ゲル濾過による目的タンパク質の分子量の測定は、目的タンパク質の分配係数 \(\mathrm{K_{av}}\) を分子量既知のタンパク質から得られた \(\mathrm{K_{av}}\) 値と比較することで行う。つまり、いくつかの分子量既知のタンパク質の \(\mathrm{K_{av}}\) 値を算出し、分子量に対して \(\mathrm{K_{av}}\) 値をプロットして検量線を描き、それを用いて目的のタンパク質の分子量を決定する。今回検量線の作製に用いるマーカーは、球状タンパク質であるThyroglobulin、Catalase、Albumin 及び Chymotrypsinogen A である。標準タンパク質としては、HMW Gel Filtration Calibration Kit(GE Healthcare)などが市販されている。分子量を決定するためには、用いるマーカーの範囲内に目的タンパク質が溶出されなければならない。そのため、目的タンパク質の溶出体積が下で用いる標準タンパク質の範囲外の場合には、それ以外の標準タンパク質を選ぶ必要がある。以下、具体的な実験の手順について記述する。サンプルの一例として、Rad51B タンパク質の分子量測定について記述した。Rad51B タンパク質は350アミノ酸からなる分子量38257 Da、Theoretical pI 5.79値のタンパク質である。
装置・器具・試薬
- Superdex 200 HR10/30(GE Healthcare)
直径 1 cm × 高さ 30 cm - AKTAexplorer 10S(GE Healthcare)
- タンパク質低吸着シリンジフィルター
(例)MILLEX-GV Syringe Driven Filter Unit フィルター材質:親水性 PVDF フィルター孔径:0.22 μm フィルター直径:4 mm(MILLIPORE) - バッファー用メンブレンフィルターユニット
(例)Vaccuum Driven Disposable Filtration System フィルター孔径:0.22 μm 容量:500 ml(IWAKI)
1)カラムの平衡化
上述した方法と同様、まず 1.2 CV のランニングバッファーを用いてカラムを平衡化する(流速 0.5 ml/min で約1時間)。分子量を測定する際には、サンプルの溶けているバッファーと同様の組成のバッファーをランニングバッファーとして用いる。また、1 ml のサンプルループを接続し、蒸留水でよく洗浄した後に、サンプルループ内もランニングバッファーに平衡化しておく。
ランニングバッファーの一例
- 20 mM Sodium Phosphate(pH 7.2)
- 150 mM NaCl
- 0.1 mM EDTA
- 10% glycerol
- 2 mM 2-mercaptoethanol
2)排除体積の決定と標準タンンパク質の溶出
排除体積を測定するために Blue Dextran 2000 を用いる。まず、Blue Dextran 2000(1 mg/ml, 300 μl)をランニングバッファーに溶解する。0.22 μM のフィルターにかけて不溶解物を除く。サンプルループに 250 μl のサンプルを添加し、1.2 CV のランニングバッファーによりサンプルを溶出する。この際、サンプルの添加量(empty loop)は 1 ml に設定する。溶出終了後、再び 1.2 CV のランニングバッファーを用いてカラムを平衡化する。
次に、
- Thyroglobulin 2 mg/ml MW 669,000
- Catalase 5 mg/ml MW 232,000
- Albumin 7 mg/ml MW 67,000
- Chymotrypsinogen A 3 mg/ml MW 25,000
(MW = Molecular Weight)
を 300 μl のランニングバッファーに溶解し、フィルターにかけて不溶解物を除く。サンプルループに 250 μl のサンプルを添加し、先程と同様の方法でサンプルを溶出する。この際、流速も同じ速さにする。溶出終了後、再び 1.2 CV のランニングバッファーを用いてカラムを平衡化する。
3)サンプルの溶出
予めフィルターにかけた 250 μl のサンプルをサンプルループに添加し、1.2 CV のランニングバッファーによりサンプルを溶出する。この際も、流速はこれまでと同じ速さで行なう。溶出後は、前述した方法でカラムを洗浄する。
例として Rad51B タンパク質を添加した際の結果を(図1)に示す。
4)検量線の作製と分子量の決定
得られた3つのデータから、それぞれのタンパク質の溶出体積(溶出ピークの中心までの溶出液の体積)を測定する(UNICORN software 内の peak integrate より自動的に測定できる)。以下の式に溶出体積を代入し、\(\mathrm{K_{av}}\) 値を計算する。
\[ \mathrm{K_{av}} = \frac{\text{(溶出体積)} - \text{(排除体積)}}{\text{(ベッド体積)} - \text{(排除体積)}} \]
\(\mathrm{K_{av}}\) 値に対して分子量を対数値でプロットして、検量線を作製する。最後に、目的タンパク質の溶出体積から \(\mathrm{K_{av}}\) 値を計算し、検量線から分子量を決定する。これらの計算は、Microsoft Excel を用いて行っている。まず、分子量既知のタンパク質から得られた \(\mathrm{K_{av}}\) 値を用いて、横軸を分子量(対数値)、縦軸を \(\mathrm{K_{av}}\) 値とした散布図を作成する。次にグラフに近似曲線を追加する。この際、オプションでグラフに数式を表示するように設定する。上述した標準タンパク質により得られた結果から作成した検量線を(図2)に示した。このようにして得た数式に目的タンパク質の \(\mathrm{K_{av}}\) 値を代入し、分子量を算出する。但し、目的のタンパク質の形状が標準タンパク質の形状(上述したマーカーは球状)と大きく異なる場合には、算出される目的タンパク質の分子量には大きなずれが生じる。そのため、どのような構造をとるか未知のタンパク質の場合には、ゲル濾過により算出された分子量はあくまで目安であることを認知しておく必要がある。例えば、(図1)に示した Rad51B タンパク質の溶出体積から、(図2)の検量線を用いて分子量を算出すると 50729 Da となる。
工夫とコツ
1)ランニングバッファーの組成
ランニングバッファーは分離能には影響しないが、pH やイオン強度、変性剤や界面活性剤の有無はタンパク質の立体構造やタンパク質サブユニット間の相互作用に変化を及ぼす。そのため、これらを考慮して目的のたんぱく質に合ったバッファー条件を十分検討する必要がある。また、カラムに吸着しやすいものもあるので、そのような場合にも塩濃度などのバッファー条件の検討が必要である。さらに、吸着しやすいサンプルを溶出した際は、カラムを NaOH で十分に洗浄し、吸着したタンパク質を除去しておくことがカラムの分離能の維持に重要である。
また、粘性の高いバッファーはカラム内の圧力を高めるため、高濃度の glycerol 条件下(30%以上)で行う際には、流速が遅くなり溶出にとても時間がかかることに注意する必要がある。
2)溶出されたサンプルの性質
ゲル濾過によりタンパク質の精製を行う際、目的タンパク質のピークがシングルピークではなく、肩がでたり2つのピークに分かれたりした場合には、タンパク質の有する活性が異なる恐れがあるので、得られたフラクションはまとめずにそれぞれを解析することが重要である。たとえシングルピークであった場合でも、最初はまとめずにそれぞれのフラクションを解析することをお奨めする。先に溶出されたサンプルと後のほうで溶出されたサンプルでは SDS-PAGE 上では同じような精製度でも、有する活性の強弱が異なることは十分ありうる。
文献
- アマシャム バイオサイエンス株式会社, Gel Filtration Principles and Methods 日本語版 (2003)
- ロバート K. スコープス, 新・タンパク質精製法 理論と実際, 213–224, シュプリンガー・フェアラーク東京株式会社 (1995)
-
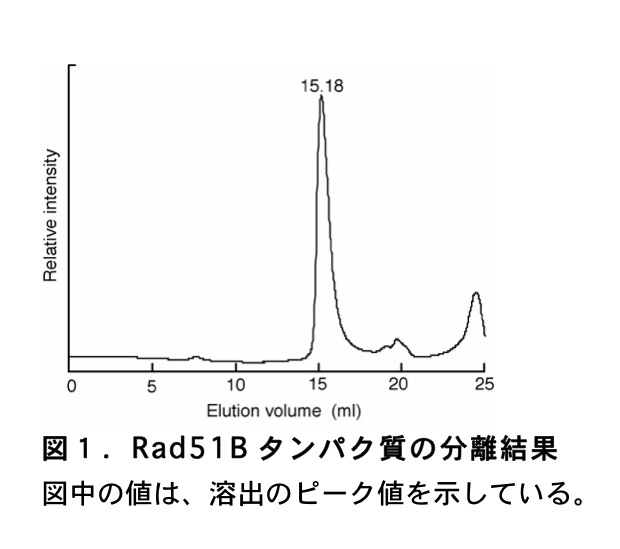
図1: -
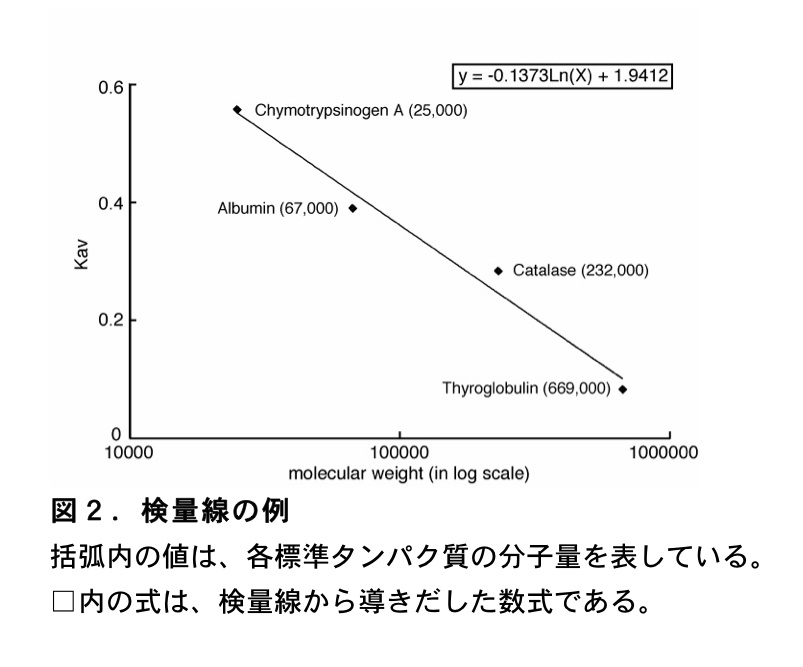
図2:
